SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 284,200 new cases of breast cancer will be diagnosed in 2021 and about 44,130 individuals will die of the disease, largely due to metastatic recurrence.
Breast cancer is heterogeneous malignancy and using global gene expression analyses, 6 breast cancer intrinsic subtypes have been established. They include Luminal A, Luminal B, HER2-enriched, Claudin-low, Basal-like, and a Normal breast-like group. Triple Negative Breast Cancer (TNBC) is a heterogeneous, molecularly diverse group of breast cancers and are ER (Estrogen Receptor), PR (Progesterone Receptor) and HER2 (Human Epidermal Growth Factor Receptor-2) negative. TNBC accounts for 15-20% of invasive breast cancers, with a higher incidence noted in young patients and African American females. It is a surrogate for the inherently aggressive Basal-like breast cancer subtype. This group has the worse prognosis compared to other breast cancer subtypes and is usually aggressive, and tumors tend to be high grade. Patients with TNBC are at a higher risk for both local and distant recurrence and often develop visceral metastases. Those with metastatic disease have one of the worst prognoses of all cancers, with a median Overall Survival of 13 months. The majority of patients with TNBC who develop metastatic disease do so within the first 3 years after diagnosis, whereas those without recurrence during this period of time have survival rates similar to those with ER-positive breast cancers. Basal-like breast cancer subtype is also a marker of hereditary breast cancer susceptibility. Multiparity may increase the risk of TNBC and reduce the likelihood of developing ER-positive breast cancer. The lack of known recurrent oncogenic drivers in patients with metastatic TNBC, presents a major therapeutic challenge.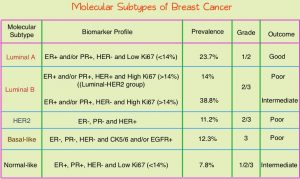
Nonetheless, patients with TNBC often receive chemotherapy in the neoadjuvant, adjuvant or metastatic settings and approximately 30-40% of patients achieve a pathological Complete Response (pCR) in the neoadjuvant setting. In addition to increasing the likelihood of tumor resectability and breast preservation, patients achieving a pCR following neoadjuvant chemotherapy have a longer Event Free Survival (EFS) and Overall Survival (OS). Those who do not achieve a pathological Complete Response tend to have a poor prognosis. For all these reasons, pCR is considered a valid endpoint for clinical testing of neoadjuvant therapy in patients with early stage TNBC. Patients with TNBC are at a high risk for recurrence if they have residual invasive disease, following completion of standard neoadjuvant chemotherapy.
In the Phase III CREATE-X trial, the addition of adjuvant XELODA® (Capecitabine) therapy was found to be safe and effective in prolonging Disease Free Survival (DFS) and Overall Survival (OS) among patients with HER2-negative breast cancer, who had residual invasive disease on pathological evaluation, following neoadjuvant chemotherapy (N Engl J Med 2017;376:2147-2159).
Based on the preclinical models supporting the use of platinum agents in the TNBC Basal-like subtype, the EA1131 trial was conducted to test the hypothesis that adjuvant platinum chemotherapy would improve invasive DFS compared with XELODA®, in patients with clinical Stage II-III TNBC, who had Basal-like subtype invasive residual disease in the breast, following neoadjuvant chemotherapy. The aim of this study was to assess whether platinum chemotherapy would be as effective, or more effective than XELODA® (noninferiority design with superiority alternative – Hybrid design).
In this study, 410 patients with clinical Stage II or III TNBC who had completed at least one full cycle of taxane with or without anthracycline-containing neoadjuvant chemotherapy were randomly assigned to receive XELODA® 1000 mg/m2 orally twice daily, days 1-14, every 3 weeks, for a total of six cycles, or a platinum agent (treating physician choice of Cisplatin 75 mg/m2 or Carboplatin AUC 6 on day 1) IV, once every 3 weeks, for a total of four cycles. Radiation Therapy before or after study treatment completion, was required for all patients after breast-conservation surgery. Postmastectomy Radiation Therapy was required for patients with primary tumors more than 5 cm or those with 4 or more positive axillary lymph nodes. TNBC subtype (Basal versus non-Basal) was determined by PAM50 in the residual disease. The Primary end point was invasive DFS (time from random assignment to the earliest disease recurrence, invasive contralateral cancer, second primary cancer, or death) in patients with Basal subtype TNBC.
After a recent interim analysis, the Data and Safety Monitoring Committee recommended stopping the trial, as it was unlikely that further follow up would show noninferiority or superiority of platinum chemotherapy. After a median follow up of 20 months, the 3-year invasive DFS among the 308 patients with Basal subtype TNBC for platinum chemotherapy was 42% versus 49% for XELODA®. Further, Grade 3 and 4 toxicities were more common in the platinum chemotherapy group. The 3-year Relapse Free Survival as well as Overall Survival was also in favor of XELODA® group versus Platinum group. There was no benefit noted with platinum chemotherapy in any of the subsets of randomized patients.
It was concluded from this study that platinum agents do not improve outcomes in patients with Basal subtype TNBC, who have residual disease following neoadjuvant chemotherapy, and are associated with more severe toxicities, when compared with XELODA®. All participants in this study had a lower than expected 3-year invasive DFS regardless of study treatment, highlighting the need for better therapies in this high-risk population. The authors added that these study findings have an immediate impact in clinical practice, and adjuvant use of platinum agents in this patient population should only be considered in the context of a clinical trial.
Randomized Phase III Postoperative Trial of Platinum-Based Chemotherapy Versus Capecitabine in Patients With Residual Triple-Negative Breast Cancer Following Neoadjuvant Chemotherapy: ECOG-ACRIN EA1131. Mayer IA, Zhao F, Arteaga CL, et al. DOI: 10.1200/JCO.21.00976 Journal of Clinical Oncology. Published online June 06, 2021.