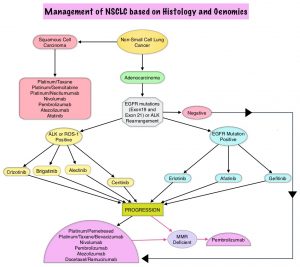
SUMMARY: Lung cancer is the second most common cancer in both men and women and accounts for about 13% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2017 about 222,500 new cases of lung cancer will be diagnosed and over 155,000 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of Non Small Cell Lung Cancer (NSCLC), 25% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large cell carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer. Approximately 10% to 15% of Caucasian patients and 50% of Asian patients with Adenocarcinomas, harbor activating EGFR (Epidermal Growth Factor Receptor) mutations and 90% of these mutations are either Exon 19 deletions or L858R point mutations in Exon 21. EGFR Tyrosine Kinase Inhibitors (TKIs) such as TARCEVA® (Erlotinib), IRESSA® (Gefitinib) and GILOTRIF® (Afatinib), have demonstrated a 60% to 70% response rate as monotherapy when administered as first line treatment, in patients with metastatic NSCLC, who harbor the sensitizing EGFR mutations.
Dacomitinib is a potent, irreversible, second-generation EGFR Tyrosine Kinase Inhibitor and inhibits three members of the ErbB protein family, including EGFR/HER1, HER2 and HER4. Based on the encouraging clinical activity of Dacomitinib in treatment naïve patients with advanced NSCLC, harboring activating EGFR mutations, in a phase II study (The Lancet Oncology 2014;15:1433-1441), the authors conducted a randomized phase III trial, comparing Dacomitinib with IRESSA®, as first line therapy in this patient population . This study (ARCHER 1050) randomized 452 patients in a 1:1 ratio to either receive Dacomitinib 45 mg PO daily (N=227) or IRESSA® 250 mg PO daily (N=225). Eligible patients had newly diagnosed stage IIIB/IV or recurrent NSCLC, harboring an activating EGFR mutation (Exon 19 deletions or L858R point mutations in Exon 21, with or without Exon 20 T790M mutations). Treatments groups were well balanced and patients were stratified by race and EGFR mutation subtype. The Primary endpoint was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR) and Duration of Response (DoR).
The median PFS for patients who received Dacomitinib was 14.7 months compared with 9.2 months for the group who received IRESSA® (HR=0.59; P<0.0001). This meant a 41% reduction in the risk of cancer progression or death with Dacomitinib compared with IRESSA®. The median Duration of Response was 14.8 months with Dacomitinib versus 8.3 months with IRESSA® (HR= 0.40; P<0.0001). As expected, patients in the Dacomitinib group experienced more side effects such as skin rash and diarrhea and this has been attributed to the stronger suppression of the EGFRs in the normal healthy tissues.
The authors concluded that ARCHER 1050 is the first phase III trial comparing EGFR TKIs head-to-head, and this study demonstrated clinically meaningful superiority of Dacomitinib, when compared to IRESSA®, in treatment naïve NSCLC patients, with activating EGFR mutations. Further, the PFS achieved with Dacomitinib in this study is among the highest observed, when compared with other EGFR Tyrosine Kinase Inhibitors, for this cancer type. Dacomitinib versus gefitinib for the first-line treatment of advanced EGFR mutation positive non-small cell lung cancer (ARCHER 1050): A randomized, open-label phase III trial. Mok T, Cheng Y, Zhou X, et al. J Clin Oncol 35, 2017 (suppl; abstr LBA9007)