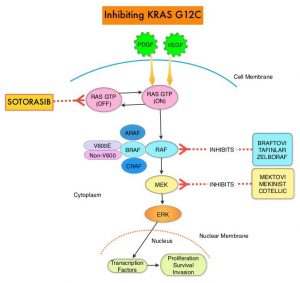
SUMMARY: The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. According to the US Lung Cancer Mutation Consortium (LCMC), two thirds of patients with advanced adenocarcinoma of the lung, have a molecular driver abnormality. The most common oncogenic drivers in patients with advanced adenocarcinoma of the lung are, KRAS in 25%, EGFR in 21% and ALK in 8%, as well as other mutations in BRAF, MET, HER2, AKT1 and fusions involving RET and ROS oncogenes. These mutations are mutually exclusive, and the presence of two simultaneous mutations, are rare.
The ASCO and Ontario Health (Cancer Care Ontario) NSCLC Expert Panel updated the 2017 ASCO guideline on systemic therapy for patients with Stage IV NSCLC with driver alterations and provided evidence-based recommendations, based on a systematic review of Randomized Controlled Trials (RCTs) from December 2015 to January 2020 and meeting abstracts from ASCO 2020.
This clinical practice guideline addresses three comprehensive clinical questions for patients with Stage IV NSCLC with driver alterations
1) What is the most effective first-line therapy?
2) What is the most effective second-line therapy?
3) Is there a role for a third-line therapy or beyond?
The guideline addresses patients with NSCLC in the following histologic or subgroups: EGFR, ALK, ROS1, BRAF, MET, RET, HER2, and NTRK. This update does not apply to patients with Stage IV NSCLC without known driver alterations and those with rarer histologies such as large cell, neuroendocrine, etc.
Summary of Key Recommendations
Recommendation 1.1: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with T790M, L858R, or exon 19 deletion mutations, Osimertinib should be offered.
Recommendations 1.2, 1.3, 1.4, and 1.5: For patients with Stage IV NSCLC and driver alterations in EGFR-if Osimertinib is not available
֍In the first-line setting, if Osimertinib is not available, Gefitinib with chemotherapy may be offered or Dacomitinib may be offered.
֍Other options that may be offered include Afatinib or Erlotinib/Bevacizumab or Erlotinib/Ramucirumab or Gefitinib, Erlotinib, or Icotinib.
Recommendation 1.6: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with a Performance Status (PS) of 3, an EGFR Tyrosine Kinase Inhibitor (TKI) may be offered.
Recommendation 1.7: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with EGFR mutations other than exon 20 insertion mutations, T790M, L858R, or exon 19 deletion alterations, Afatinib may be offered or Osimertinib may be offered or treatments outlined in the ASCO/OH nondriver mutation guideline may be offered.
Recommendation 1.8: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the first-line setting, for patients with any activating EGFR mutation (including exon 20 insertion mutations), regardless of Programmed Death Ligand-1 (PD-L1) expression levels, single-agent immunotherapy should not be used.
Recommendation 1.9: For patients with Stage IV NSCLC and driver alterations in EGFR causing resistance to first- and second-generation EGFR TKIs
֍In the first-line setting, for patients with EGFR exon 20 insertion mutation causing resistance to first- and second-generation EGFR TKIs, doublet chemotherapy with or without Bevacizumab or standard treatment outlined in the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 2.1 and 2.2: For patients with Stage IV NSCLC and driver alterations in EGFR
֍In the second-line setting, for patients who did not receive Osimertinib and have a T790M mutation at the time of progressive disease, Osimertinib should be offered.
֍In the second-line setting, for patients with any EGFR mutation who have progressed on EGFR TKIs with no T790M mutation OR whose disease has progressed on Osimertinib, treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendation 3.1: For patients with Stage IV NSCLC and driver alterations in ALK
֍In the first-line setting, Alectinib or Brigatinib should be offered.
֍In the first-line setting, if Alectinib and Brigatinib are not available, Ceritinib or Crizotinib should be offered.
Recommendations 4.1, 4.2, and 4.3: For patients with stage IV NSCLC and driver alterations in ALK
֍In the second-line setting, if Alectinib or Brigatinib was given in the first-line setting, Lorlatinib may be offered.
֍In the second-line setting, if Crizotinib was given in the first-line setting, then Alectinib, Brigatinib, or Ceritinib should be offered.
֍In the third-line setting, if Crizotinib was given in the first-line setting and Alectinib, Brigatinib, or Ceritinib in the second-line setting, then Lorlatinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 5.1, 5.2, and 5.3: For patients with Stage IV NSCLC and driver alterations in ROS1
֍In the first-line setting, Crizotinib or Entrectinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered or Ceritinib or Lorlatinib may be offered.
Recommendations 6.1 and 6.2: For patients with Stage IV NSCLC and driver alterations in ROS1
֍In the second-line setting, if ROS1-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
֍In the second-line setting, if nontargeted therapy was given in the first-line setting, Crizotinib, Ceritinib, or Entrectinib may be offered.
Recommendations 7.1 and 7.2: For patients with Stage IV NSCLC and driver alterations with BRAF V600E mutation
֍In the first-line setting, Dabrafenib/Trametinib may be offered or standard first-line treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 8.1, 8.2 and 8.3: For patients with Stage IV NSCLC and driver alterations with BRAF V600E mutation
֍In the second-line setting, if previous BRAF/MEK-targeted therapy (Dabrafenib/Trametinib) was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
֍In the second-line setting, if BRAF-targeted therapy was not given in the first-line setting, Dabrafenib/Trametinib may be offered or Dabrafenib or Vemurafenib alone may be offered.
֍If previous chemotherapy, immunotherapy, and/or BRAF-targeted therapy were given in the first- or subsequent-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
Recommendation 8.4: For patients with Stage IV NSCLC and driver alterations with BRAF mutations other than V600E
֍In the second-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
Recommendations 9.1 and 9.2: For patients with Stage IV NSCLC and MET exon 14 skipping mutation
֍In the first-line setting, for patients with an MET exon 14 skipping mutation, MET-targeted therapy with Capmatinib or Tepotinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 10.1 and 10.2: For patients with Stage IV NSCLC and MET exon 14 skipping mutation
֍In the second-line setting, for MET abnormalities other than exon 14 skipping mutations or if MET-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline should be offered.
֍In the second-line setting, patients with an MET exon 14 skipping mutation who previously received or were ineligible for first-line chemotherapy with or without immunotherapy (ie. if MET-targeted therapy was not given in the first-line setting), Capmatinib or Tepotinib may be offered.
Recommendations 11.1, 11.2, and 11.3: For patients with Stage IV NSCLC and driver alterations in RET
֍In the first-line setting, Selpercatinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered or Pralsetinib may be offered.
Recommendations 12.1, 12.2, and 12.3: For patients with Stage IV NSCLC and driver alterations in RET
֍In the second-line setting, if RET-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
֍In the second-line setting, if RET-targeted therapy was not given in the first-line setting, Selpercatinib may be offered or Pralsetinib may be offered.
Recommendations 13.1 and 13.2: For patients with Stage IV NSCLC and driver alterations in NTRK
֍In the first-line setting, Entrectinib or Larotrectinib may be offered or standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
Recommendations 14.1 and 14.2: For patients with Stage IV NSCLC and driver alterations in NTRK
֍In the second-line setting, if NTRK-targeted therapy was given in the first-line setting, standard treatment based on the ASCO/OH nondriver mutation guideline may be offered.
֍In the second-line setting, if NTRK-targeted therapy was not given in the first-line setting, Entrectinib or Larotrectinib may be offered.
Therapy for Stage IV Non–Small-Cell Lung Cancer With Driver Alterations: ASCO and OH (CCO) Joint Guideline Update. Hanna NH, Robinson AG, Temin S, et al. J Clin Oncol. 2021;39: 1040-1091