
The FDA on December 14, 2023, approved WELIREG® for patients with advanced Renal Cell Carcinoma (RCC) following a Programmed Death receptor-1 (PD-1) or Programmed Death-Ligand 1 (PD-L1) inhibitor and a Vascular Endothelial Growth Factor Tyrosine Kinase Inhibitor (VEGF-TKI). WELIREG® is a product of Merck & Co., Inc.
Tag: Renal Cell Carcinoma
FDA Approves WELIREG® for Advanced Renal Cell Carcinoma
SUMMARY: The FDA on December 14, 2023, approved Belzutifan (WELIREG®) for patients with advanced Renal Cell Carcinoma (RCC) following a Programmed Death receptor-1 (PD-1) or Programmed Death-Ligand 1 (PD-L1) inhibitor and a Vascular Endothelial Growth Factor Tyrosine Kinase Inhibitor (VEGF-TKI). The American Cancer Society estimates that 81,800 new cases of kidney and renal pelvis cancers will be diagnosed in the United States in 2023 and about 14,890 people will die from this disease. Clear cell Renal Cell Carcinoma (RCC) is by far the most common type of kidney cancer in adults. Modifiable risk factors include smoking, obesity, workplace exposure to certain substances and high blood pressure. The five-year survival of patients with advanced RCC is about 14% and there is a significant need for improved therapies for this disease.
Patients with advanced RCC are often treated with immune checkpoint inhibitors and Vascular Endothelial Growth Factor Receptor (VEGFR) targeted Tyrosine Kinase Inhibitors, either in combination or sequentially. However upon progression on these therapies, there are limited treatment options and there is an unmet medical need.
The VHL (Von Hippel-Lindau) protein is a tumor suppressor gene located on the short arm of chromosome 3p. It is frequently mutated and inactivated in approximately 90% of clear cell Renal Cell Carcinomas (ccRCC). The VHL gene under normal conditions binds to Hypoxia-Inducible Factors (HIFs) and facilitates degradation of this factor. Under hypoxic conditions and in patients having biallelic loss of function and mutation of VHL genes, HIFs are not degraded. High HIF levels and subsequent overproduction of VEGF, PDGF and TGF-alpha, resulting in increased angiogenesis, increased tumor cell proliferation and survival, as well as metastasis.
Belzutifan (WELIREG®) is a a first-in-class, oral, HIF-2alfa inhibitor approved in the US for adult patients with Von Hippel-Lindau (VHL) disease who require therapy for associated Renal Cell Carcinoma (RCC), Central Nervous System (CNS) Hemangioblastomas, or Pancreatic NeuroEndocrine Tumors (pNET), not requiring immediate surgery. This approval was based on the Overall Response Rate (ORR) and Duration of Response (DOR) data from the Phase II LITESPARK-004 trial.
The present FDA approval was based on LITESPARK-005, which is a randomized, open-label, Phase III trial, in which Belzutifan was compared with Everolimus in pretreated advanced Renal Cell Carcinoma (RCC). In this study, 746 enrolled patients with metastatic clear cell RCC whose disease progressed after treatment with both an immune checkpoint inhibitor, such as a PD-1 or PD-L1 inhibitor, and VEGF-TKI, in sequence or in combination, were randomly assigned 1:1 to receive either Belzutifan 120 mg orally daily (N=374) or Everolimus 10 mg orally daily (N=372), until disease progression or unacceptable toxicity. The dual Primary endpoints were Progression Free Survival (PFS) by Blinded Independent Central Review (BICR) and Overall Survival (OS). Secondary endpoints included Overall Response Rate (ORR) by BICR and Safety.
At the first pre-specified interim analysis at a median follow up of 18.4 months, Belzutifan significantly reduced the risk of disease progression or death by 25% compared to Everolimus (HR=0.75; P<0.001). The results at the second pre-specified interim analysis were consistent with first interim analysis. At a median follow-up of 25.7 months, Belzutifan significantly reduced the risk of disease progression or death by 26% compared to Everolimus (HR=0.74; P<0.001). The estimated 12-month PFS rate was 33.7% for patients who received Belzutifan versus 17.6% for patients who received Everolimus, and the estimated 18-month PFS rate was 22.5% and 9.0%, respectively. The Overall Survival data favored Belzutifan compared to Everolimus at both the first and second interim analysis, but did not reach statistical significance and will be tested at a subsequent analysis.
There was a statistically significant improvement in ORR at both the first and second interim analysis, and the ORR was 22.7% with a Complete Response rate 3.5% for patients who received Belzutifan versus an ORR of 3.5% with no patients achieving a Complete Response for patients who received Everolimus (P<0.00001). The time to response with Belzutifan was about three months. Quality of Life favored Belzutifan.
Treatment-related adverse events and in particular Grade 3 adverse events were similar in both treatment groups. Adverse events leading to treatment discontinuation occurred in 5.9% of patients who received Belzutifan and 14.7% among those who received Everolimus. The most common side effects associated with Belzutifan were anemia, fatigue, nausea, constipation, peripheral edema, dyspnea and arthralgia.
It was concluded that Belzutifan was associated with a statistically significant improvement in Progression Free Survival and Overall Response Rate compared to Everolimus in patients with advanced clear cell Renal Cell Carcinoma, after immune checkpoint and anti-angiogenic therapies. They added that this is the first Phase III trial to show positive results in advanced RCC following standard therapies and the first drug with a new mechanism of action to demonstrate efficacy in this group of patients.
Belzutifan versus everolimus in participants (pts) with previously treated advanced clear cell renal cell carcinoma (ccRCC): Randomized open-label phase III LITESPARK-005 study. Albiges L, Rini BI, Peltola K, et al. DOI:https://doi.org/10.1016/j.annonc.2023.10.090. LBA88
Late Breaking Abstract – ESMO 2023: WELIREG® Significantly Improves Outcomes in Advanced Pretreated Clear Cell Renal Cell Carcinoma
SUMMARY: The American Cancer Society estimates that 81,800 new cases of kidney and renal pelvis cancers will be diagnosed in the United States in 2023 and about 14,890 people will die from this disease. Clear cell Renal Cell Carcinoma (RCC) is by far the most common type of kidney cancer in adults. Modifiable risk factors include smoking, obesity, workplace exposure to certain substances and high blood pressure. The five-year survival of patients with advanced RCC is about 14% and there is a significant need for improved therapies for this disease.
Patients with advanced RCC are often treated with immune checkpoint inhibitors and Vascular Endothelial Growth Factor Receptor (VEGFR) targeted Tyrosine Kinase Inhibitors, either in combination or sequentially. However upon progression on these therapies, there are limited treatment options and there is an unmet medical need.
The VHL (Von Hippel-Lindau) protein is a tumor suppressor gene located on the short arm of chromosome 3p. It is frequently mutated and inactivated in approximately 90% of clear cell Renal Cell Carcinomas (ccRCC). The VHL gene under normal conditions binds to Hypoxia-Inducible Factors (HIFs) and facilitates degradation of this factor. Under hypoxic conditions and in patients having biallelic loss of function and mutation of VHL genes, HIFs are not degraded. High HIF levels and subsequent overproduction of VEGF, PDGF and TGF-alpha, resulting in increased angiogenesis, increased tumor cell proliferation and survival, as well as metastasis.
Belzutifan (WELIREG®) is a a first-in-class, oral, HIF-2alfa inhibitor approved in the US for adult patients with Von Hippel-Lindau (VHL) disease who require therapy for associated Renal Cell Carcinoma (RCC), Central Nervous System (CNS) Hemangioblastomas, or Pancreatic NeuroEndocrine Tumors (pNET), not requiring immediate surgery. This approval was based on the Overall Response Rate (ORR) and Duration of Response (DOR) data from the Phase II LITESPARK-004 trial.
LITESPARK-005 is a randomized, open-label, Phase III trial in which Belzutifan was compared with Everolimus in pretreated advanced ccRCC. In this study, 746 enrolled patients with metastatic clear cell renal cell carcinoma whose disease progressed after treatment with both an immune checkpoint inhibitor, such as a PD-1 or PD-L1 inhibitor, and VEGF-TKI, in sequence or in combination, were randomly assigned 1:1 to receive either Belzutifan 120 mg orally daily (N=374) or Everolimus 10 mg orally daily (N=372), until disease progression or unacceptable toxicity. The dual Primary endpoints were Progression Free Survival (PFS) by Blinded Independent Central Review (BICR) and Overall Survival (OS). Secondary endpoints included Overall Response Rate (ORR) by BICR and Safety.
At the first pre-specified interim analysis at a median follow up of 18.4 months, Belzutifan significantly reduced the risk of disease progression or death by 25% compared to Everolimus (HR=0.75; P<0.001). The results at the second pre-specified interim analysis were consistent with first interim analysis. At a median follow-up of 25.7 months, Belzutifan significantly reduced the risk of disease progression or death by 26% compared to Everolimus (HR=0.74; P<0.001). The estimated 12-month PFS rate was 33.7% for patients who received Belzutifan versus 17.6% for patients who received Everolimus, and the estimated 18-month PFS rate was 22.5% and 9.0%, respectively. The Overall Survival data favored Belzutifan compared to Everolimus at both the first and second interim analysis, but did not reach statistical significance and will be tested at a subsequent analysis.
There was a statistically significant improvement in ORR at both the first and second interim analysis, and the ORR was 22.7% with a Complete Response rate 3.5% for patients who received Belzutifan versus an ORR of 3.5% with no patients achieving a Complete Response for patients who received Everolimus (P<0.00001). The time to response with Belzutifan was about three months. Quality of Life favored Belzutifan.
Treatment-related adverse events and in particular Grade 3 adverse events were similar in both treatment groups. Adverse events leading to treatment discontinuation occurred in 5.9% of patients who received Belzutifan and 14.7% among those who received Everolimus. The most common side effects associated with Belzutifan were anemia, fatigue, nausea, constipation, peripheral edema, dyspnea and arthralgia.
It was concluded that Belzutifan was associated with a statistically significant improvement in Progression Free Survival and Overall Response Rate compared to Everolimus in patients with advanced clear cell Renal Cell Carcinoma, after immune checkpoint and anti-angiogenic therapies. They added that this is the first Phase III trial to show positive results in advanced RCC following standard therapies and the first drug with a new mechanism of action to demonstrate efficacy in this group of patients.
Belzutifan versus everolimus in participants (pts) with previously treated advanced clear cell renal cell carcinoma (ccRCC): Randomized open-label phase III LITESPARK-005 study. Albiges L, Rini BI, Peltola K, et al. DOI:https://doi.org/10.1016/j.annonc.2023.10.090. LBA88
Review of 44-month follow-up data and primary analysis for cabozantinib + nivolumab in 1L aRCC
Written by Thomas Hutson, D.O., Pharm. D.
Sponsored by Exelixis, Inc.
At the 2023 GU Cancer Symposium jointly sponsored by ASCO, ASTRO, and SUO, Dr Maurice Burotto presented the minimum 3-year follow-up efficacy and safety data for the ITT population in the Phase 3 CheckMate-9ER trial. These extended follow-up analysis results continue to support cabozantinib + nivolumab as a first-line treatment for patients with advanced RCC.1,2
CheckMate‐9ER was a randomized (1:1), open‐label, Phase 3 trial vs sunitinib in 651 patients with previously untreated aRCC with a clear‐cell component.1,3
- Dosing: cabozantinib 40 mg (starting dose) PO once daily in combination with nivolumab 240 mg flat dose IV every 2 weeks vs sunitinib 50 mg (starting dose) PO once daily for 4 weeks, followed by 2 weeks off, per cycle.1
- The starting dose of cabozantinib is 40 mg when used in combination with nivolumab, unlike the 60‐mg recommended starting dose for single‐agent therapy1
- Endpoints assessed1,3-6:
- Primary endpoint was PFS*
- Secondary endpoints were OS, ORR,* and safety
- Quality of life: evaluated as an exploratory endpoint using the FKSI‐19 scale, and the clinical significance of the results is unknown
- Additional exploratory endpoints: biomarkers, pharmacokinetics, immunogenicity, and PFS‐2
- Updated efficacy analysis: conducted when 271 events were observed based on the pre‐specified number of events for the pre‐planned final analysis of OS1,7,8
- Patient population in CheckMate-9ER was representative of the aRCC population seen in clinical practice (cabozantinib + nivolumab, n=323; sunitinib, n=328)1,3,4,9-11
- IMDC risk categories3
- Favorable: 23% of cabozantinib + nivolumab patients; 22% of sunitinib patients
- Intermediate: 58% of cabozantinib + nivolumab patients; 57% of sunitinib patients
- Poor: 19% of cabozantinib + nivolumab patients; 21% of sunitinib patients
- Prior nephrectomy: 69% of cabozantinib + nivolumab patients; 71% of sunitinib patients9
- Liver metastases: 23% of cabozantinib + nivolumab patients; 16% of sunitinib patients3
- Bone metastases: 24% of cabozantinib + nivolumab patients; 22% of sunitinib patients3
- ≥2 metastatic sites: 80% of cabozantinib + nivolumab patients; 78% of sunitinib patients3
- IMDC risk categories3
Primary analysis results (median follow‐up time of 18.1 months; range: 10.6‐30.6 months)3:
- Median PFS was 16.6 months for cabozantinib + nivolumab (95% CI: 12.5-24.9; n=323) compared with 8.3 months for sunitinib (95% CI: 7.0-9.7); HR=0.51 (95% CI: 0.41-0.64)1*
- Probability of OS at 12 months was 85.7% for cabozantinib + nivolumab (95% CI: 81.3-89.1) compared with 75.6% for sunitinib (95% CI: 70.5-80.0); HR=0.60 (98.89% CI: 0.40-0.89). The median OS was not reached in either group3
- ORR was 55.7% for cabozantinib + nivolumab (95% CI: 50.1-61.2; n=323) compared with 27.1% for sunitinib (95% CI: 22.4-32.3; n=328)1*
Updated pre-planned analysis of OS (median follow‐up: 32.9 months; range: 25.4‐45.4 months)1,7,8:
- Median OS was 37.7 months for cabozantinib + nivolumab (95% CI: 35.5‐NR; n=323) compared with 34.3 months for sunitinib (95% CI: 29.0‐NR; n=328); HR=0.70 (95% CI: 0.55‐0.90)
More than 2 years of additional follow-up from the primary analysis, which makes it the longest available for CheckMate-9ER (median follow‐up: 44 months; range: 36.5-56.5 months):2
- Median PFS was 16.6 months for cabozantinib + nivolumab (95% CI: 12.8-19.5; n=323) compared with 8.4 months for sunitinib (95% CI: 7.0-9.7; n=328); HR=0.59 (95% CI: 0.49-0.71)2
- ORR was 56.0% for cabozantinib + nivolumab (95% CI: 50.4-61.5; n=323) compared with 28.0% for sunitinib (95% CI: 23.3-33.2; n=328)2
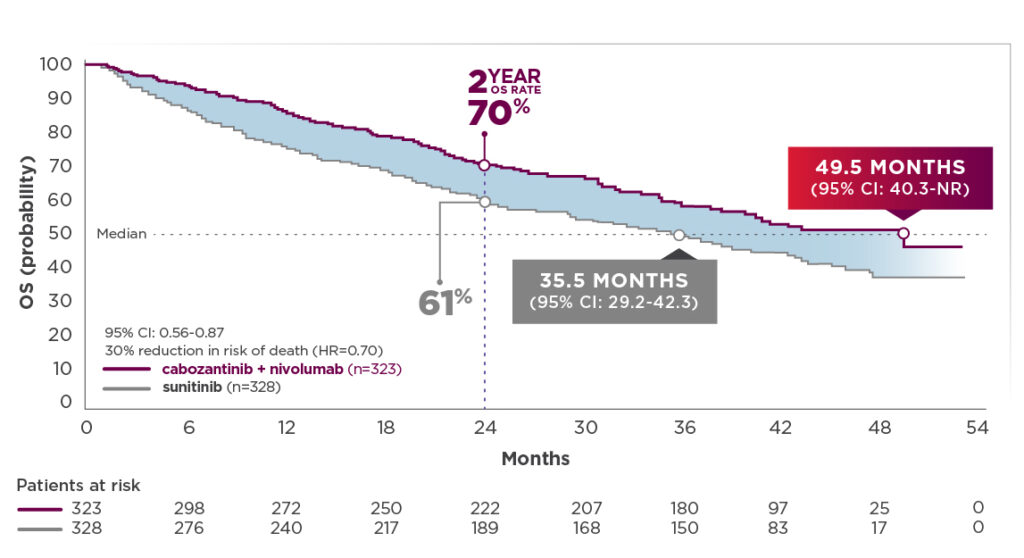
- Median OS (depicted in graph below) was 49.5 months for cabozantinib + nivolumab (95% CI: 40.3-NR; n=323) compared with 35.5 months for sunitinib (95% CI: 29.2-42.3); HR=0.70 (95% CI: 0.56-0.87)2,12
- The median OS for cabozantinib + nivolumab patients was 14 months longer than the sunitinib arm.2,12 This substantial difference was derived without adjustment for subsequent cancer therapy3
44-month follow-up analysis2,12
OS: Median follow-up time of 44.0 months; range: 36.5-56.5 months
PFS and ORR results in patients with bone, liver, and/or lung metastasis shown below13:
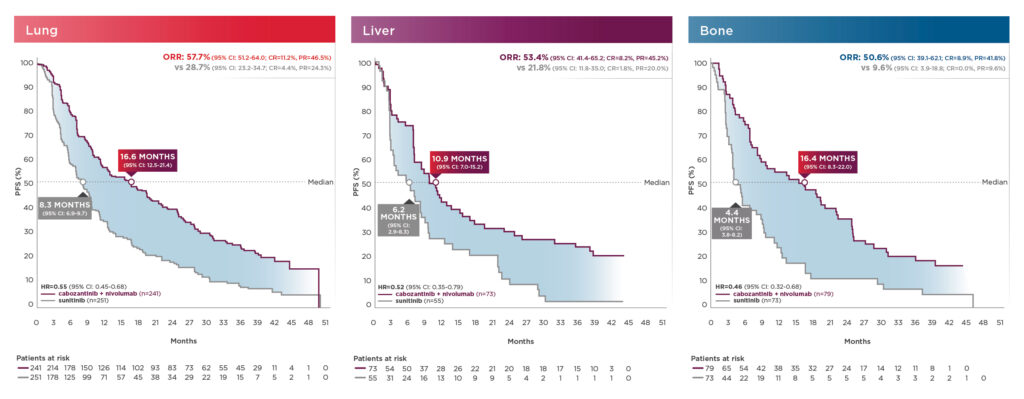
These exploratory analyses are descriptive in nature. Subgroups were not powered to show differences between treatment arms, and results should be considered hypothesis generating.7
In CheckMate-9ER, serious adverse reactions occurred in 48% of patients receiving cabozantinib + nivolumab.1 Serious adverse reactions reported in ≥2% of patients were diarrhea, pneumonia, pneumonitis, pulmonary embolism, urinary tract infection, and hyponatremia.1 Fatal intestinal perforations occurred in 3 (0.9%) patients.1 The most common adverse reactions (≥20%) in patients receiving cabozantinib + nivolumab (n=320) vs sunitinib (n=320) were diarrhea (64% vs 47%), fatigue (51% vs 50%), hepatotoxicity (44% vs 26%), palmar‐plantar erythrodysesthesia (40% vs 41%), stomatitis (37% vs 46%), rash (36% vs 14%), hypertension (36% vs 39%), hypothyroidism (34% vs 30%), musculoskeletal pain (33% vs 29%), decreased appetite (28% vs 20%), nausea (27% vs 31%), dysgeusia (24% vs 22%), abdominal pain (22% vs 15%), upper respiratory tract infection (20% vs 8%), and cough (20% vs 17%).1
- Cabozantinib may be interrupted or reduced due to adverse events to 20 mg daily or 20 mg every other day.1 The average dosage of cabozantinib in CheckMate-9ER was 30 mg14
- If previously receiving 20 mg once every other day, resume at same dosage. If not tolerated, discontinue cabozantinib1
- Adverse reactions leading to discontinuation of either cabozantinib or nivolumab occurred in 20% of patients, which included 8% for only cabozantinib and 7% for only nivolumab.3 It is important to note that 6% of patients in the CheckMate‐9ER trial discontinued both cabozantinib and nivolumab at the same time due to adverse events, compared with 17% of patients in the sunitinib arm who permanently discontinued their treatment3
- Cabozantinib should be permanently discontinued for Grade 3 or 4 hemorrhage, development of a GI perforation or Grade 4 fistula, acute myocardial infarction or Grade 2 or higher cerebral infarction, Grade 3 or 4 arterial thromboembolic events or Grade 4 venous thromboembolic events, Grade 4 hypertension/hypertensive crisis or Grade 3 hypertension/hypertensive crisis that cannot be controlled, nephrotic syndrome, or reversible posterior leukoencephalopathy syndrome1
- For patients being treated with cabozantinib + nivolumab, if ALT or AST >10x ULN or >3x ULN with concurrent total bilirubin ≥2x ULN, both cabozantinib + nivolumab should be permanently discontinued1
In summary, the 44-month follow-up data indicate that:
- After a minimum follow-up of 3 years, survival and response benefits were maintained with cabozantinib + nivolumab, showing consistent outcomes as in previous follow-ups1,2
- Median OS with cabozantinib + nivolumab improved by 11.8 months since the previous follow-up analysis. Median OS with cabozantinib + nivolumab was 49.5 months compared with 35.5 months for sunitinib1,2,7,8,12
- The median OS for cabozantinib + nivolumab patients was substantially longer (14 months) than the sunitinib arm. This difference was derived without adjustment for subsequent cancer therapy2,3
- No new safety signals emerged with additional follow-up in either arm2
- Among patients treated with cabozantinib + nivolumab, the discontinuation rate due to ARs to cabozantinib alone was 10% and nivolumab alone was 10%, vs 11% for sunitinib.2 This may allow patients receiving cabozantinib + nivolumab to stay on therapy and thus allow them to achieve efficacy benefits
Dr Hutson received a fee for participating in this program, and his comments reflect his opinions and are not intended to constitute medical advice for individual patients.
[Footnotes]
*PFS and ORR were assessed by BICR.1
1L=first‐line; ALT=alanine aminotransferase; AR=adverse reaction; aRCC=advanced RCC; ASCO=American Society of Clinical Oncology; AST=aspartate aminotransferase; ASTRO=American Society for Radiation Oncology; BICR=blinded independent central review; CI=confidence interval; CR=complete response; FKSI‐19=Functional Assessment of Cancer Therapy‐Kidney Symptom Index 19; HR=hazard ratio; IMDC=International Metastatic RCC Database Consortium; IV=intravenous; NR=not reached; ORR=objective response rate; OS=overall survival; PFS=progression‐free survival; PFS‐2=PFS after subsequent therapy; PO=by mouth; PR=partial response; RCC=renal cell carcinoma; SUO=Society of Urologic Oncology; ULN=upper limit of normal.
INDICATIONS
CABOMETYX® (cabozantinib) is indicated for the treatment of patients with advanced renal cell carcinoma (RCC).
CABOMETYX, in combination with nivolumab, is indicated for the first-line treatment of patients with advanced RCC.
CABOMETYX is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib.
CABOMETYX is indicated for the treatment of adult and pediatric patients 12 years of age and older with locally advanced or metastatic differentiated thyroid cancer (DTC) that has progressed following prior VEGFR-targeted therapy and who are radioactive iodine-refractory or ineligible.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Hemorrhage: Severe and fatal hemorrhages occurred with CABOMETYX. The incidence of Grade 3 to 5 hemorrhagic events was 5% in CABOMETYX patients in RCC, HCC, and DTC studies. Discontinue CABOMETYX for Grade 3 or 4 hemorrhage and prior to surgery as recommended. Do not administer CABOMETYX to patients who have a recent history of hemorrhage, including hemoptysis, hematemesis, or melena.
Perforations and Fistulas: Fistulas, including fatal cases, occurred in 1% of CABOMETYX patients. Gastrointestinal (GI) perforations, including fatal cases, occurred in 1% of CABOMETYX patients. Monitor patients for signs and symptoms of fistulas and perforations, including abscess and sepsis. Discontinue CABOMETYX in patients who experience a Grade 4 fistula or a GI perforation.
Thrombotic Events: CABOMETYX increased the risk of thrombotic events. Venous thromboembolism occurred in 7% (including 4% pulmonary embolism) and arterial thromboembolism in 2% of CABOMETYX patients. Fatal thrombotic events occurred in CABOMETYX patients. Discontinue CABOMETYX in patients who develop an acute myocardial infarction or serious arterial or venous thromboembolic events that require medical intervention.
Hypertension and Hypertensive Crisis: CABOMETYX can cause hypertension, including hypertensive crisis. Hypertension was reported in 37% (16% Grade 3 and <1% Grade 4) of CABOMETYX patients. Do not initiate CABOMETYX in patients with uncontrolled hypertension. Monitor blood pressure regularly during CABOMETYX treatment. Withhold CABOMETYX for hypertension that is not adequately controlled with medical management; when controlled, resume at a reduced dose. Permanently discontinue CABOMETYX for severe hypertension that cannot be controlled with anti-hypertensive therapy or for hypertensive crisis.
Diarrhea: Diarrhea occurred in 62% of CABOMETYX patients. Grade 3 diarrhea occurred in 10% of CABOMETYX patients. Monitor and manage patients using antidiarrheals as indicated. Withhold CABOMETYX until improvement to ≤ Grade 1, resume at a reduced dose.
Palmar-Plantar Erythrodysesthesia (PPE): PPE occurred in 45% of CABOMETYX patients. Grade 3 PPE occurred in 13% of CABOMETYX patients. Withhold CABOMETYX until improvement to Grade 1 and resume at a reduced dose for intolerable Grade 2 PPE or Grade 3 PPE.
Hepatotoxicity: CABOMETYX in combination with nivolumab can cause hepatic toxicity with higher frequencies of Grades 3 and 4 ALT and AST elevations compared to CABOMETYX alone.
Monitor liver enzymes before initiation of and periodically throughout treatment. Consider more frequent monitoring of liver enzymes than when the drugs are administered as single agents. For elevated liver enzymes, interrupt CABOMETYX and nivolumab and consider administering corticosteroids.
With the combination of CABOMETYX and nivolumab, Grades 3 and 4 increased ALT or AST were seen in 11% of patients. ALT or AST >3 times ULN (Grade ≥2) was reported in 83 patients, of whom 23 (28%) received systemic corticosteroids; ALT or AST resolved to Grades 0-1 in 74 (89%). Among the 44 patients with Grade ≥2 increased ALT or AST who were rechallenged with either CABOMETYX (n=9) or nivolumab (n=11) as a single agent or with both (n=24), recurrence of Grade ≥2 increased ALT or AST was observed in 2 patients receiving CABOMETYX, 2 patients receiving nivolumab, and 7 patients receiving both CABOMETYX and nivolumab. Withhold and resume at a reduced dose based on severity.
Adrenal Insufficiency: CABOMETYX in combination with nivolumab can cause primary or secondary adrenal insufficiency. For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold CABOMETYX and/or nivolumab and resume CABOMETYX at a reduced dose depending on severity.
Adrenal insufficiency occurred in 4.7% (15/320) of patients with RCC who received CABOMETYX with nivolumab, including Grade 3 (2.2%), and Grade 2 (1.9%) adverse reactions. Adrenal insufficiency led to permanent discontinuation of CABOMETYX and nivolumab in 0.9% and withholding of CABOMETYX and nivolumab in 2.8% of patients with RCC.
Approximately 80% (12/15) of patients with adrenal insufficiency received hormone replacement therapy, including systemic corticosteroids. Adrenal insufficiency resolved in 27% (n=4) of the 15 patients. Of the 9 patients in whom CABOMETYX with nivolumab was withheld for adrenal insufficiency, 6 reinstated treatment after symptom improvement; of these, all (n=6) received hormone replacement therapy and 2 had recurrence of adrenal insufficiency.
Proteinuria: Proteinuria was observed in 8% of CABOMETYX patients. Monitor urine protein regularly during CABOMETYX treatment. For Grade 2 or 3 proteinuria, withhold CABOMETYX until improvement to ≤ Grade 1 proteinuria, resume CABOMETYX at a reduced dose. Discontinue CABOMETYX in patients who develop nephrotic syndrome.
Osteonecrosis of the Jaw (ONJ): ONJ occurred in <1% of CABOMETYX patients. ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain, or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to CABOMETYX initiation and periodically during treatment. Advise patients regarding good oral hygiene practices. Withhold CABOMETYX for at least 3 weeks prior to scheduled dental surgery or invasive dental procedures, if possible. Withhold CABOMETYX for development of ONJ until complete resolution, resume at a reduced dose
Impaired Wound Healing: Wound complications occurred with CABOMETYX. Withhold CABOMETYX for at least 3 weeks prior to elective surgery. Do not administer CABOMETYX for at least 2 weeks after major surgery and until adequate wound healing. The safety of resumption of CABOMETYX after resolution of wound healing complications has not been established.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS): RPLS, a syndrome of subcortical vasogenic edema diagnosed by characteristic findings on MRI, can occur with CABOMETYX. Evaluate for RPLS in patients presenting with seizures, headache, visual disturbances, confusion, or altered mental function. Discontinue CABOMETYX in patients who develop RPLS.
Thyroid Dysfunction: Thyroid dysfunction, primarily hypothyroidism, has been observed with CABOMETYX. Based on the safety population, thyroid dysfunction occurred in 19% of patients treated with CABOMETYX, including Grade 3 in 0.4% of patients.
Patients should be assessed for signs of thyroid dysfunction prior to the initiation of CABOMETYX and monitored for signs and symptoms of thyroid dysfunction during CABOMETYX treatment. Thyroid function testing and management of dysfunction should be performed as clinically indicated.
Hypocalcemia: CABOMETYX can cause hypocalcemia. Based on the safety population, hypocalcemia occurred in 13% of patients treated with CABOMETYX, including Grade 3 in 2% and Grade 4 in 1% of patients. Laboratory abnormality data were not collected in CABOSUN.
In COSMIC-311, hypocalcemia occurred in 36% of patients treated with CABOMETYX, including Grade 3 in 6% and Grade 4 in 3% of patients.
Monitor blood calcium levels and replace calcium as necessary during treatment. Withhold and resume at reduced dose upon recovery or permanently discontinue CABOMETYX depending on severity.
Embryo-Fetal Toxicity: CABOMETYX can cause fetal harm. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Verify the pregnancy status of females of reproductive potential prior to initiating CABOMETYX and advise them to use effective contraception during treatment and for 4 months after the last dose.
ADVERSE REACTIONS
The most common (≥20%) adverse reactions are:
CABOMETYX as a single agent: diarrhea, fatigue, PPE, decreased appetite, hypertension, nausea, vomiting, weight decreased, constipation.
CABOMETYX in combination with nivolumab: diarrhea, fatigue, hepatotoxicity, PPE, stomatitis, rash, hypertension, hypothyroidism, musculoskeletal pain, decreased appetite, nausea, dysgeusia, abdominal pain, cough, and upper respiratory tract infection.
DRUG INTERACTIONS
Strong CYP3A4 Inhibitors: If coadministration with strong CYP3A4 inhibitors cannot be avoided, reduce the CABOMETYX dosage. Avoid grapefruit or grapefruit juice.
Strong CYP3A4 Inducers: If coadministration with strong CYP3A4 inducers cannot be avoided, increase the CABOMETYX dosage. Avoid St. John’s wort.
USE IN SPECIFIC POPULATIONS
Lactation: Advise women not to breastfeed during CABOMETYX treatment and for 4 months after the final dose.
Hepatic Impairment: In patients with moderate hepatic impairment, reduce the CABOMETYX dosage. Avoid CABOMETYX in patients with severe hepatic impairment.
Please see accompanying full Prescribing Information by clicking here.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.
References
- CABOMETYX® (cabozantinib) Prescribing Information. Exelixis, Inc.
- Burotto M, Powles T, Escudier B, et al. Nivolumab plus cabozantinib versus sunitinib for first-line treatment of advanced renal cell carcinoma: 3-year follow-up from the phase 3 CheckMate 9ER trial. Poster presented at Cancer Immunotherapy and Immunomonitoring Conference; April 24-27, 2023.
- Choueiri TK, Powles T, Burotto M, et al; CheckMate 9ER Investigators. Nivolumab plus cabozantinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2021;384(9):829‐841.
- Motzer RJ, Choueiri TK, Powles T, et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal cell carcinoma: outcomes by sarcomatoid histology and updated trial results with extended follow‐up of CheckMate 9ER. Poster presented at Genitourinary Cancers Symposium; February 11‐13, 2021
- Choueiri TK, Powles T, Burotto M, et al; CheckMate 9ER Investigators. Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma [supplementary appendix]. N Engl J Med. 2021;384(9):829-841.
- Choueiri TK, Powles T, Burotto M, et al; CheckMate 9ER Investigators. Nivolumab plus cabozantinib versus sunitinib for advanced renal‐cell carcinoma [protocol]. N Engl J Med. 2021;384(9):829‐841.
- Motzer RJ, Powles T, Burotto M, et al. Nivolumab plus cabozantinib versus sunitinib in first‐line treatment for advanced renal cell carcinoma (CheckMate 9ER): long‐term follow‐up results from an open‐label, randomized, phase 3 trial. Lancet Oncol. 2022;23(7):888‐898.
- Powles T, Choueiri TK, Burotto M, et al. Final overall survival analysis and organ‐specific target lesion assessments with 2‐year follow‐up in CheckMate 9ER: nivolumab plus cabozantinib versus sunitinib for patients with advanced renal cell carcinoma. Poster presented at the American Society of Clinical Oncology Genitourinary Cancers Symposium; February 17‐19, 2022
- Data on file. Topline 9ER. Exelixis, Inc.
- Savard M-F, Wells JC, Graham J, et al. Real-world assessment of clinical outcomes among first-line sunitinib patients with clear cell metastatic renal cell carcinoma (mRCC) by the International mRCC Database Consortium risk group. Oncologist. 2020;25(5):422-430.
- Heng DYC, Xie W, Regan MM, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol. 2009;27(34):5794-5799.
- Data on file. Exelixis, Inc.
- Data on file. Exelixis, Inc.
- Data on file. Final Clinical Study Report for Study CA2099ER. Bristol Myers Squibb.
©2023 Exelixis, Inc. CA‐2644-1 07/23
OPDIVO® and the related logo are registered trademarks of Bristol‐Myers Squibb Company
Clinical considerations in 1L advanced renal cell carcinoma (aRCC)
Written by Manojkumar Bupathi, MD, MS
Sponsored by Exelixis
The treatment of patients with aRCC is evolving rapidly, with new regimens being developed and approved for 1L therapy. When choosing a regimen for patients, there are a number of treatment components to assess, including but not limited to the patient’s clinical presentation, their ability to tolerate treatment, and potential impact on quality of life. In my practice, I look at the location of metastases in the patient, whether they have symptomatic disease, and whether treatment‐related adverse reactions can be managed with supportive care.
A 1L aRCC treatment I consider is the FDA‐approved combination of CABOMETYX® (cabozantinib) + OPDIVO® (nivolumab). I believe CABOMETYX + OPDIVO offers a balance of data including superior OS, safety and tolerability, and patient‐ reported QoL data.1‐3*
CheckMate‐9ER was a randomized (1:1), open‐label, Phase 3 trial vs sunitinib in 651 patients with previously untreated aRCC with a clear‐cell component.1,2,4
- Dosing: CABOMETYX 40 mg (starting dose) PO once daily in combination with OPDIVO 240 mg flat dose IV every 2 weeks vs sunitinib 50 mg (starting dose) PO once daily for 4 weeks, followed by 2 weeks off, per cycle.1
- Starting dose: unlike the 60‐mg recommended starting dose for single‐ agent therapy, the starting dose of CABOMETYX is 40 mg when used in combination with OPDIVO1
- Primary endpoint: PFS1
- Secondary endpoints: OS, ORR, and safety1,4
- Quality of life: evaluated as an exploratory endpoint using the FKSI‐19 scale, and the clinical significance of the results is unknown2,3
- Additional exploratory endpoints: biomarkers, pharmacokinetics, immunogenicity, and PFS‐22,5
- Updated efficacy analysis: conducted when 271 events were observed based on the pre‐specified number of events for the pre‐planned final analysis of OS1,6
Primary analysis results (median follow‐up time of 18.1 months; range: 10.6‐30.6 months)2:
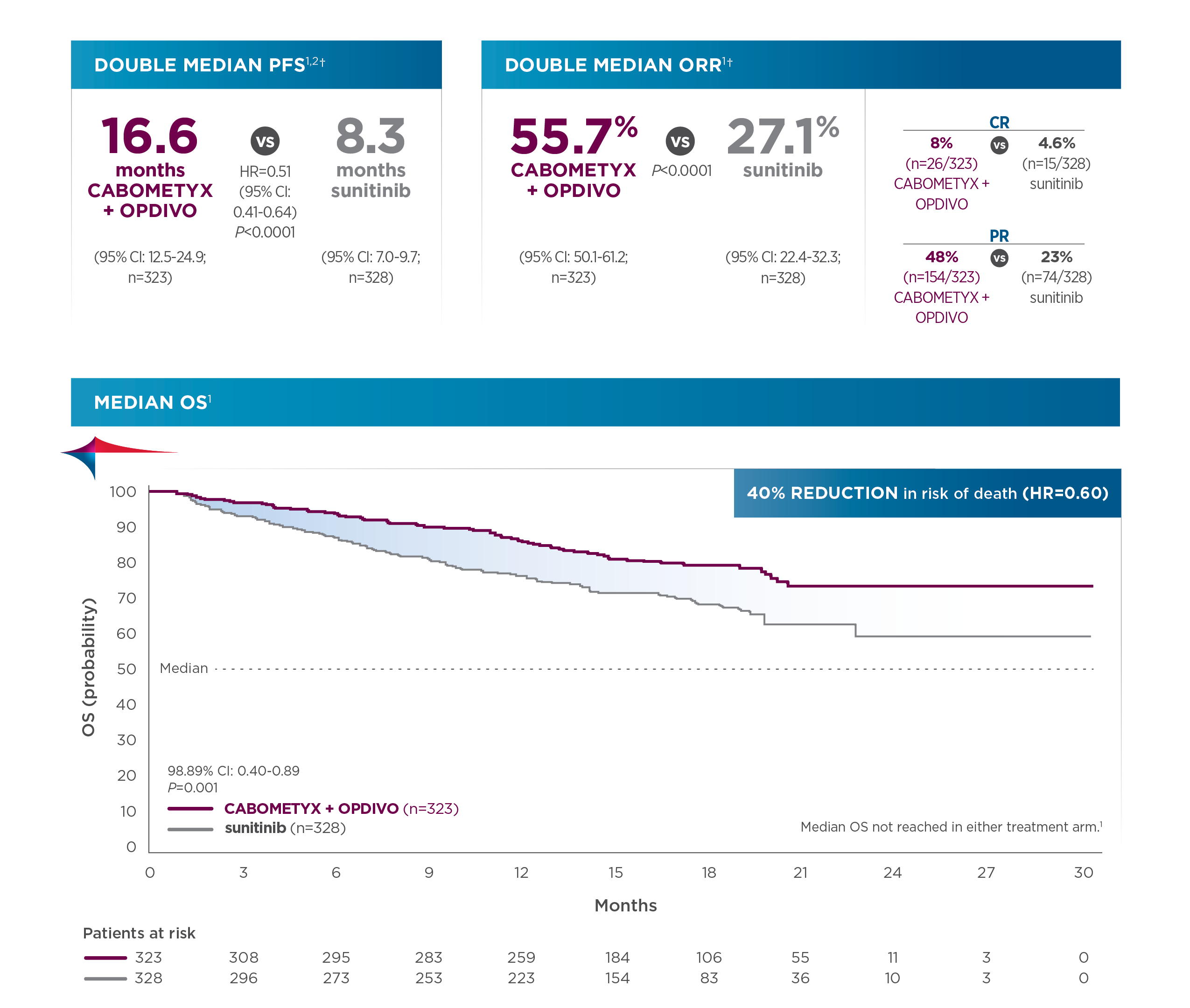
Updated analysis of OS (median follow‐up: 32.9 months; range: 25.4‐45.4 months):
- Median OS was 37.7 months for CABOMETYX + OPDIVO (95% CI: 35.5‐NR; n=323) compared with 34.3 months for sunitinib (95% CI: 29.0‐NR; n=328); HR=0.70 (95% CI: 0.55‐0.90).1,6,7
When selecting a 1L treatment for my patients with aRCC, I review all the efficacy endpoints along with tolerability and safety data, as well as dosing. In Checkmate 9ER, serious adverse reactions occurred in 48% of patients receiving CABOMETYX+ OPDIVO (n=320).1 Serious adverse reactions reported in ≥2% of patients were diarrhea, pneumonia, pneumonitis, pulmonary embolism, urinary tract infection, and hyponatremia.1 Fatal intestinal perforations occurred in 3 (0.9%) patients.1 The most common adverse reactions (≥20%) in patients receiving CABOMETYX + OPDIVO were diarrhea (64%), fatigue (51%), hepatotoxicity (44%), palmar‐plantar erythrodysesthesia syndrome (40%), stomatitis (37%), rash (36%), hypertension (36%), hypothyroidism (34%), musculoskeletal pain (33%), decreased appetite (28%), nausea (27%), dysgeusia (24%), abdominal pain (22%), upper respiratory tract infection (20%), and cough (20%).1
- CABOMETYX may be interrupted or reduced due to adverse events to 20 mg daily or 20 mg every other day.1
- If previously receiving 20 mg once every other day, resume at same dosage. If not tolerated, discontinue CABOMETYX.1
- Adverse reactions leading to discontinuation of either CABOMETYX or OPDIVO occurred in 20% of patients, which included 8% (CABOMETYX only) and 7% (OPDIVO only). It is important to note that 6% of patients in the CheckMate‐9ER trial discontinued both CABOMETYX and OPDIVO due to adverse events, compared with 16.9% of patients in the sunitinib arm who permanently discontinued their treatment.1,8
- CABOMETYX should be permanently discontinued for Grade 3 or 4 hemorrhage, development of a GI perforation or Grade 4 fistula, acute myocardial infarction or Grade 2 or higher cerebral infarction, Grade 3 or 4 arterial thromboembolic events or Grade 4 venous thromboembolic events, Grade 4 hypertension/hypertensive crisis or Grade 3 hypertension/hypertensive crisis that cannot be controlled, nephrotic syndrome, or reversible posterior leukoencephalopathy syndrome1
- For patients being treated with CABOMETYX in combination with OPDIVO, if ALT or AST >10x ULN or >3x ULN with concurrent total bilirubin ≥2x ULN, both CABOMETYX and OPDIVO should be permanently discontinued1
With these data, I feel comfortable using CABOMETYX + OPDIVO as a first‐line treatment for appropriate aRCC patients. I’d like to add that, according to the NCCN guidelines, CABOMETYX + OPDIVO is a category 1 preferred regimen in clear cell aRCC, which gives me additional confidence to prescribe this regimen for appropriate patients.9‡
Dr Bupathi received a fee for participating in this program, and his comments reflect his opinions and are not intended to constitute medical advice for individual patients.
[Footnotes]
*Superior OS vs sunitinib in patients with previously untreated aRCC. Primary analysis OS results: 40% reduction in risk of death with CABOMETYX + OPDIVO vs sunitinib (HR=0.60 [98.89% CI: 0.40‐0.89]; P=0.001); median OS was not reached in either arm. The primary endpoint was PFS, and secondary endpoints included OS, ORR, and safety. Quality of life was evaluated as an exploratory endpoint using the FKSI‐19 scale, and the clinical significance is unknown.1-3
†PFS and ORR were assessed by BICR.1
‡The trial population size of CheckMate‐9ER was 651 patients.1
1L=first‐line; ALT=alanine aminotransferase; AST=aspartate aminotransferase; BICR=blinded independent central review; CI=confidence interval; FDA=Food and Drug Administration; FKSI‐19=Functional Assessment of Cancer Therapy‐Kidney Symptom Index 19; HR=hazard ratio; IO=immunotherapy; IV=intravenous; NR=not reached; ORR=objective response rate; OS=overall survival; PFS=progression‐free survival; PFS‐2=PFS after subsequent therapy; PO=by mouth; QoL=quality of life; TKI=tyrosine kinase inhibitor; ULN=upper limit of normal.
INDICATIONS
CABOMETYX® (cabozantinib), in combination with nivolumab, is indicated for the first‐line treatment of patients with advanced renal cell carcinoma (RCC).
CABOMETYX is indicated for the treatment of patients with advanced RCC.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Hemorrhage: Severe and fatal hemorrhages occurred with CABOMETYX. The incidence of Grade 3 to 5 hemorrhagic events was 5% in CABOMETYX patients in RCC, HCC, and DTC studies. Discontinue CABOMETYX for Grade 3 or 4 hemorrhage and prior to surgery as recommended. Do not administer CABOMETYX to patients who have a recent history of hemorrhage, including hemoptysis, hematemesis, or melena.
Perforations and Fistulas: Fistulas, including fatal cases, occurred in 1% of CABOMETYX patients. Gastrointestinal (GI) perforations, including fatal cases, occurred in 1% of CABOMETYX patients. Monitor patients for signs and symptoms of fistulas and perforations, including abscess and sepsis. Discontinue CABOMETYX in patients who experience a Grade 4 fistula or a GI perforation.
Thrombotic Events: CABOMETYX increased the risk of thrombotic events. Venous thromboembolism occurred in 7% (including 4% pulmonary embolism) and arterial thromboembolism in 2% of CABOMETYX patients. Fatal thrombotic events occurred in CABOMETYX patients. Discontinue CABOMETYX in patients who develop an acute myocardial infarction or serious arterial or venous thromboembolic events that require medical intervention.
Hypertension and Hypertensive Crisis: CABOMETYX can cause hypertension, including hypertensive crisis. Hypertension was reported in 37% (16% Grade 3 and <1% Grade 4) of CABOMETYX patients. Do not initiate CABOMETYX in patients with uncontrolled hypertension. Monitor blood pressure regularly during CABOMETYX treatment. Withhold CABOMETYX for hypertension that is not adequately controlled with medical management; when controlled, resume at a reduced dose. Permanently discontinue CABOMETYX for severe hypertension that cannot be controlled with anti‐hypertensive therapy or for hypertensive crisis.
Diarrhea: Diarrhea occurred in 62% of CABOMETYX patients. Grade 3 diarrhea occurred in 10% of CABOMETYX patients. Monitor and manage patients using antidiarrheals as indicated. Withhold CABOMETYX until improvement to ≤ Grade 1, resume at a reduced dose.
Palmar‐Plantar Erythrodysesthesia (PPE): PPE occurred in 45% of CABOMETYX patients. Grade 3 PPE occurred in 13% of CABOMETYX patients. Withhold CABOMETYX until improvement to Grade 1 and resume at a reduced dose for intolerable Grade 2 PPE or Grade 3 PPE.
Hepatotoxicity: CABOMETYX in combination with nivolumab can cause hepatic toxicity with higher frequencies of Grades 3 and 4 ALT and AST elevations compared to CABOMETYX alone.
Monitor liver enzymes before initiation of and periodically throughout treatment. Consider more frequent monitoring of liver enzymes than when the drugs are administered as single agents. For elevated liver enzymes, interrupt CABOMETYX and nivolumab and consider administering corticosteroids.
With the combination of CABOMETYX and nivolumab, Grades 3 and 4 increased ALT or AST were seen in 11% of patients. ALT or AST >3 times ULN (Grade ≥2) was reported in 83 patients, of whom 23 (28%) received systemic corticosteroids; ALT or AST resolved to Grades 0‐1 in 74 (89%). Among the 44 patients with Grade ≥2 increased ALT or AST who were rechallenged with either CABOMETYX (n=9) or nivolumab (n=11) as a single agent or with both (n=24), recurrence of Grade ≥2 increased ALT or AST was observed in 2 patients receiving CABOMETYX, 2 patients receiving nivolumab, and 7 patients receiving both CABOMETYX and nivolumab.
Withhold and resume at a reduced dose based on severity.
Adrenal Insufficiency: CABOMETYX in combination with nivolumab can cause primary or secondary adrenal insufficiency. For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold CABOMETYX and/or nivolumab and resume CABOMETYX at a reduced dose depending on severity.
Adrenal insufficiency occurred in 4.7% (15/320) of patients with RCC who received CABOMETYX with nivolumab, including Grade 3 (2.2%), and Grade 2 (1.9%) adverse reactions. Adrenal insufficiency led to permanent discontinuation of CABOMETYX and nivolumab in 0.9% and withholding of CABOMETYX and nivolumab in 2.8% of patients with RCC.
Approximately 80% (12/15) of patients with adrenal insufficiency received hormone replacement therapy, including systemic corticosteroids. Adrenal insufficiency resolved in 27% (n=4) of the 15 patients. Of the 9 patients in whom CABOMETYX with nivolumab was withheld for adrenal insufficiency, 6 reinstated treatment after symptom improvement; of these, all (n=6) received hormone replacement therapy and 2 had recurrence of adrenal insufficiency.
Proteinuria: Proteinuria was observed in 8% of CABOMETYX patients. Monitor urine protein regularly during CABOMETYX treatment. For Grade 2 or 3 proteinuria, withhold CABOMETYX until improvement to ≤ Grade 1 proteinuria, resume CABOMETYX at a reduced dose. Discontinue CABOMETYX in patients who develop nephrotic syndrome.
Osteonecrosis of the Jaw (ONJ): ONJ occurred in <1% of CABOMETYX patients. ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain, or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to CABOMETYX initiation and periodically during treatment. Advise patients regarding good oral hygiene practices. Withhold CABOMETYX for at least 3 weeks prior to scheduled dental surgery or invasive dental procedures, if possible. Withhold CABOMETYX for development of ONJ until complete resolution, resume at a reduced dose.
Impaired Wound Healing: Wound complications occurred with CABOMETYX. Withhold CABOMETYX for at least 3 weeks prior to elective surgery. Do not administer CABOMETYX for at least 2 weeks after major surgery and until adequate wound healing. The safety of resumption of CABOMETYX after resolution of wound healing complications has not been established.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS): RPLS, a syndrome of subcortical vasogenic edema diagnosed by characteristic findings on MRI, can occur with CABOMETYX. Evaluate for RPLS in patients presenting with seizures, headache, visual disturbances, confusion, or altered mental function. Discontinue CABOMETYX in patients who develop RPLS.
Thyroid Dysfunction: Thyroid dysfunction, primarily hypothyroidism, has been observed with CABOMETYX. Based on the safety population, thyroid dysfunction occurred in 19% of patients treated with CABOMETYX, including Grade 3 in 0.4% of patients.
Patients should be assessed for signs of thyroid dysfunction prior to the initiation of CABOMETYX and monitored for signs and symptoms of thyroid dysfunction during CABOMETYX treatment. Thyroid function testing and management of dysfunction should be performed as clinically indicated.
Hypocalcemia: CABOMETYX can cause hypocalcemia. Based on the safety population, hypocalcemia occurred in 13% of patients treated with CABOMETYX, including Grade 3 in 2% and Grade 4 in 1% of patients. Laboratory abnormality data were not collected in CABOSUN.
In COSMIC‐311, hypocalcemia occurred in 36% of patients treated with CABOMETYX, including Grade 3 in 6% and Grade 4 in 3% of patients.
Monitor blood calcium levels and replace calcium as necessary during treatment. Withhold and resume at reduced dose upon recovery or permanently discontinue CABOMETYX depending on severity.
Embryo‐Fetal Toxicity: CABOMETYX can cause fetal harm. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Verify the pregnancy status of females of reproductive potential prior to initiating CABOMETYX and advise them to use effective contraception during treatment and for 4 months after the last dose.
ADVERSE REACTIONS
The most common (≥20%) adverse reactions are:
CABOMETYX as a single agent: diarrhea, fatigue, PPE, decreased appetite, hypertension, nausea, vomiting, weight decreased, constipation.
CABOMETYX in combination with nivolumab: diarrhea, fatigue, hepatotoxicity, PPE, stomatitis, rash, hypertension, hypothyroidism, musculoskeletal pain, decreased appetite, nausea, dysgeusia, abdominal pain, cough, and upper respiratory tract infection.
DRUG INTERACTIONS
Strong CYP3A4 Inhibitors: If coadministration with strong CYP3A4 inhibitors cannot be avoided, reduce the CABOMETYX dosage. Avoid grapefruit or grapefruit juice.
Strong CYP3A4 Inducers: If coadministration with strong CYP3A4 inducers cannot be avoided, increase the CABOMETYX dosage. Avoid St. John’s wort.
USE IN SPECIFIC POPULATIONS
Lactation: Advise women not to breastfeed during CABOMETYX treatment and for 4 months after the final dose.
Hepatic Impairment: In patients with moderate hepatic impairment, reduce the CABOMETYX dosage. Avoid CABOMETYX in patients with severe hepatic impairment.
Please see accompanying full Prescribing Information by clicking here.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1‐800‐FDA‐1088.
References
1. CABOMETYX® (cabozantinib) Prescribing Information. Exelixis, Inc, 2022.
2. Choueiri TK, Powles T, Burotto M, et al; CheckMate 9ER Investigators. Nivolumab plus cabozantinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2021;384(9):829‐841.
3. Choueiri TK, Powles T, Burotto M, et al; CheckMate 9ER Investigators. Nivolumab plus cabozantinib versus sunitinib for advanced renal‐cell carcinoma [supplementary appendix]. N Engl J Med. 2021;384(9):829‐841.
4. Motzer RJ, Choueiri TK, Powles T, et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal cell carcinoma: outcomes by sarcomatoid histology and updated trial results with extended follow‐up of CheckMate 9ER. Poster presented at Genitourinary Cancers Symposium; February 11‐ 13, 2021.
5. Choueiri TK, Powles T, Burotto M, et al; CheckMate 9ER Investigators. Nivolumab plus cabozantinib versus sunitinib for advanced renal‐cell carcinoma [protocol]. N Engl J Med. 2021;384(9):829‐841.
6. Powles T, Choueiri TK, Burotto M, et al. Final overall survival analysis and organ‐specific target lesion assessments with 2‐year follow‐up in CheckMate 9ER: nivolumab plus cabozantinib versus sunitinib for patients with advanced renal cell carcinoma. Poster presented at the American Society of Clinical Oncology Genitourinary Cancers Symposium; February 17‐19, 2022.
7. Motzer RJ, Powles T, Burotto M, et al. Nivolumab plus cabozantinib versus sunitinib in first‐line treatment for advanced renal cell carcinoma (CheckMate 9ER): long‐term follow‐up results from an open‐label, randomized, phase 3 trial. Lancet Oncol. 2022;23(7):888‐898.
8. Data on file. Final Clinical Study Report for Study CA2099ER. Bristol Myers Squibb.
9. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Kidney Cancer V.3.2023. © National Comprehensive Cancer Network, Inc. 2022.
All rights reserved. Accessed September 29, 2022. To view the most recent and complete version of the guideline, go online to NCCN.org.
©2022 Exelixis, Inc. CA‐2644 12/22
OPDIVO® and the related logo are registered trademarks of Bristol‐Myers Squibb Company.
Late Breaking Abstract – ESMO 2022: CABOMETYX®, OPDIVO® and YERVOY® in Previously Untreated Advanced Renal Cell Carcinoma
SUMMARY: The American Cancer Society estimates that 79,000 new cases of kidney cancers will be diagnosed in the United States in 2022 and about 13,920 people will die from this disease. Clear Cell Renal Cell Carcinoma (RCC) is by far the most common type of kidney cancer in adults. Modifiable risk factors include smoking, obesity, workplace exposure to certain substances and high blood pressure. The five-year survival of patients with advanced RCC is about 14% and there is a significant unmet need for improved therapies for this disease.
OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, whereas YERVOY® (Ipilimumab) is a fully human immunoglobulin G1 monoclonal antibody that blocks Immune checkpoint protein/receptor CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, also known as CD152). Blocking the Immune checkpoint proteins unleashes the T cells, resulting in T cell proliferation, activation, and a therapeutic response. The FDA in 2018, granted approvals to OPDIVO® and YERVOY® in combination, for the treatment of Intermediate or Poor-risk, previously untreated advanced Renal Cell Carcinoma.
CABOMETYX® (Cabozantinib) is an oral, small-molecule Tyrosine Kinase Inhibitor (TKI), which targets Vascular Endothelial Growth Factor Receptors (VEGFR), as well as tyrosine kinases MET and AXL. Both MET and AXL are upregulated in Renal Cell Carcinoma as a consequence of VHL inactivation, and increased expression of MET and AXL is associated with tumor progression and development of resistance to VEGFR inhibitors. Further, CABOMETYX® promotes an immune-permissive environment, which may enhance response to checkpoint inhibitors. CABOMETYX® was approved by the FDA in 2016 for the treatment of advanced Renal Cell Carcinoma.
COSMIC-313 is a global, multicenter, randomized, double-blinded, controlled, ongoing Phase III pivotal trial, conducted to evaluate the triplet combination of Cabozantinib, Nivolumab and Ipilimumab versus the doublet combination of Nivolumab and Ipilimumab, in patients with previously untreated advanced Intermediate or Poor-risk Renal Cell Carcinoma. COSMIC-313 was designed to answer whether adding Cabozantinib to dual checkpoint inhibition can improve outcomes among patients with Intermediate and Poor-risk advanced Renal Cell Carcinoma.
In this trial, 855 treatment naïve, advanced clear cell Renal Cell Carcinoma patients of IMDC (International Metastatic RCC Database Consortium) Intermediate or Poor risk were randomized 1:1 to receive Cabozantinib plus Nivolumab and Ipilimumab (N=428) or placebo plus Nivolumab and Ipilimumab (N=427). Patients in the study group received Cabozantinib 40 mg, orally once daily in combination with Nivolumab 3 mg/kg IV and Ipilimumab 1 mg/kg IV once every 3 weeks for 4 doses total followed by Cabozantinib 40 mg orally once daily and Nivolumab 480 mg/kg flat dose IV, once every 4 weeks for up to 2 years. Patients in the control group received the same regimen, but instead of Cabozantinib, received a matched placebo. Both treatment groups were well balanced. The median patient age was 60 years, 75% were men, 63% had PD-L1 expression of less than 1%, 75% had Intermediate-risk disease, 25% were Poor risk, and 65% had prior nephrectomy. The Primary endpoint was Progression Free Survival (PFS), as assessed by Blinded Independent Radiology Committee (BIRC). Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR) and Safety. The median follow up was 20.2 months.
The study met the Primary endpoint and the median PFS was not reached in the Cabozantinib group and was 11.3 months in the placebo group (HR=0.73; P=0.013). Patients treated with the Cabozantinib three-drug combination had a 27% lower risk of disease progression or death compared to those on the two drug immunotherapy combination. This PFS benefit was predominantly noted in the Intermediate-risk group. The Objective Response Rate was 43% with the Cabozantinib combination versus 36% in the placebo plus dual immunotherapy group, with 3% of patients achieving a Complete Response in both treatment groups. The Disease Control Rate was 86% and 72%, respectively. The median Duration of Response was not reached in either treatment group. Grade 3/4 adverse events occurred in 73% of patients treated with the combination of Cabozantinib, Nivolumab and Ipilimumab, and in 41% of patients treated with the Nivolumab and Ipilimumab combination. Discontinuation of all treatment agents due to adverse events occurred in 12% and 5% of patients, respectively.
The authors concluded that this is the first study to show that a TKI added to dual checkpoint inhibition significantly improved Progression Free Survival, in patients with untreated, Intermediate or Poor risk advanced kidney cancer, compared to doublet immunotherapy. Follow-up for Overall Survival is ongoing.
Phase III study of cabozantinib (C) in combination with nivolumab (N) and ipilimumab (I) in previously untreated advanced renal cell carcinoma (aRCC) of IMDC intermediate or poor risk (COSMIC-313). Choueiri TK, Powles TB, Albiges L, et al. Annals of Oncology (2022) 33 (suppl_7): S808-S869. 10.1016/annonc/annonc1089. LBA8
KEYTRUDA® (Pembrolizumab)
The FDA on November 17, 2021, approved KEYTRUDA® (Pembrolizumab) for the adjuvant treatment of patients with Renal Cell Carcinoma (RCC) at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions. KEYTRUDA® is a product of Merck & Co.
LENVIMA® (Lenvatinib) plus KEYTRUDA® (Pembrolizumab)
The FDA on August 10, 2021 approved the combination of LENVIMA® plus KEYTRUDA® for first-line treatment of adult patients with advanced Renal Cell Carcinoma (RCC). LENVIMA® is a product of Eisai Co., Ltd. and KEYTRUDA® is a product of Merck & Co.
FDA Approves LENVIMA® Plus KEYTRUDA® for Advanced Renal Cell Carcinoma
SUMMARY: The FDA on August 10, 2021, approved the combination of LENVIMA® (Lenvatinib) plus KEYTRUDA® (Pembrolizumab) for first line treatment of adult patients with advanced Renal Cell Carcinoma (RCC). The American Cancer Society estimates that 76,080 new cases of kidney cancers will be diagnosed in the United States in 2021 and about 13,780 people will die from the disease. Renal Cell Carcinoma (RCC) is by far the most common type of kidney cancer and is about twice as common in men as in women. Modifiable risk factors include smoking, obesity, workplace exposure to certain substances and high blood pressure. The five year survival of patients with advanced RCC is less than 10% and there is a significant unmet need for improved therapies for this disease.
SUTENT® (Sunitinib) is a MultiKinase Inhibitor (MKI) which simultaneously targets the tumor cell wall, vascular endothelial cell wall as well as the pericyte/fibroblast/vascular/smooth vessel cell wall, and is capable of specifically binding to tyrosine kinases inhibiting the earlier signaling events and thereby inhibits phosphorylation of VEGF receptor, PDGF receptor, FLT-3 and c-KIT. SUTENT® has been the standard first line intervention for treatment naïve patients with advanced RCC. In a large, multi-center, randomized, Phase III study, the median Progression Free Survival (PFS) with SUTENT® was 9.5 months, the Objective Response Rate (ORR) was 25%, and the median Overall Survival (OS) was 29.3 months, when compared with Interferon Alfa, in patients with treatment-naïve Renal Cell Carcinoma. This was however associated with a high rate of hematological toxicities.
KEYTRUDA® (Pembrolizumab) is a fully humanized, Immunoglobulin G4, anti-PD-1, monoclonal antibody, that binds to the PD-1 receptor and blocks its interaction with ligands PD-L1 and PD-L2. It thereby reverses the PD-1 pathway-mediated inhibition of the immune response and unleashes the tumor-specific effector T cells.
LENVIMA® (Lenvatinib) is an oral multitargeted TKI which targets Vascular Endothelial Growth Factor Receptor (VEGFR) 1-3, Fibroblast Growth Factor Receptor (FGFR) 1-4, Rearranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with antiangiogenesis properties by its ability to inhibit FGFR-1, thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting FGFR and PDGFR beta.
AFINITOR® (Everolimus) does not inhibit tyrosine kinases, but is a specific inhibitor of mTOR (Mammalian Target of Rapamycin), which is a serine/threonine kinase, normally activated further downstream in the signaling cascade. With the inhibition of mTOR, protein synthesis is inhibited resulting in decreased angiogenesis, cell proliferation and survival as well as decreased levels of HIF-1 alpha.
A combination of LENVIMA® plus AFINITOR® was shown to be associated with longer Progression Free Survival than AFINITOR® alone as second line treatment in advanced RCC (Lancet Oncol 2015;16:1473-1482). LENVIMA® plus KEYTRUDA® was shown to have promising antitumor activity in previously treated patients with RCC in a Phase IB-II trial (J Clin Oncol 2020;38:1154-1163). Based on this data, the authors conducted a multicenter, randomized, open-label, Phase III trial to compare the efficacy and safety of LENVIMA® in combination with KEYTRUDA® or AFINITOR® versus SUTENT® alone, in first line treatment of patients with advanced RCC.
The researchers randomly assigned 1069 patients with advanced RCC and no previous systemic therapy in a 1:1:1 ratio to receive LENVIMA® 20 mg orally once daily plus KEYTRUDA® 200 mg IV once every 3 weeks (N=355), LENVIMA® 18 mg orally once daily plus AFINITOR® 5 mg orally once daily (N=357) or SUTENT® 50 mg orally once daily, alternating 4 weeks on and 2 weeks off (N=357). The Primary end point was Progression Free Survival (PFS) and Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR) and Safety. The median follow up for OS was 26.6 months.
The median PFS was significantly longer with LENVIMA® plus KEYTRUDA® combination, compared to single agent SUTENT® (23.9 months versus 9.2 months, HR=0.39; P<0.001). The median PFS with the LENVIMA® plus AFINITOR® combination was also significantly longer, compared to single agent SUTENT® (14.7 months versus 9.2 months, HR=0.65; P<0.001). The PFS benefit favored the two LENVIMA® combination regimens over single agent SUTENT® across all evaluated subgroups, including those based on MSKCC prognostic risk group and International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk group. At interim analysis, the OS was significantly longer with LENVIMA® plus KEYTRUDA® than with SUTENT® (HR for death=0.66; P=0.005). This benefit was noted in most subgroups, including patients with PD-L1 positive or negative tumors, with an exception of patients with favorable risk disease as defined by IMDC criteria. Overall Survival with LENVIMA® plus AFINITOR® was however not significantly longer compared with SUTENT® (HR=1.15; P=0.30).
The confirmed ORR was 71% with LENVIMA® plus KEYTRUDA®, 53.5% with LENVIMA® plus AFINITOR®, and 36.1% with single agent SUTENT®. The Complete Response rate was 16.1% in the LENVIMA® plus KEYTRUDA® group, 9.8% in the LENVIMA® plus AFINITOR® group, and 4.2% in the SUTENT® group. The median Duration of Response in patients who had a confirmed response was 25.8 months in the LENVIMA® plus KEYTRUDA® group, 16.6 months in the LENVIMA® plus AFINITOR® group, and 14.6 months in the SUTENT® group. Grade 3 or higher Adverse Events occurred in 82.4% of the patients who received LENVIMA® plus KEYTRUDA® group, in 83.1% of the patients who received LENVIMA® plus AFINITOR®, and in 71.8% of the patients who received SUTENT®.
It was concluded that a combination of LENVIMA® plus KEYTRUDA® provided superior Progression Free Survival and Overall Survival compared to SUTENT®, in the first line treatment of patients with advanced Renal Cell Carcinoma.
Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. Motzer R, Alekseev B, Rha S-Y, et al. for the CLEAR Trial Investigators. N Engl J Med 2021; 384:1289-1300
Late Breaking Abstract – ASCO 2021: Adjuvant KEYTRUDA® Improves Disease Free Survival in Renal Cell Carcinoma
SUMMARY: The American Cancer Society estimates that 76,080 new cases of kidney cancers will be diagnosed in the United States in 2021 and about 13,780 people will die from the disease. Renal Cell Carcinoma (RCC) is by far the most common type of kidney cancer and is about twice as common in men as in women. Modifiable risk factors include smoking, obesity, workplace exposure to certain substances and high blood pressure. The five year survival of patients with advanced RCC is less than 10% and there is a significant unmet need for improved therapies for this disease.
The prognosis for patients with Renal Cell Carcinoma (RCC) is dependent on the stage of disease and risk factors. Two validated models, the University of California Los Angeles Integrated Staging System (UISS) and the Stage, Size, Grade, and Necrosis (SSIGN) score were developed, to assess the risk for relapse. UISS is based on ECOG Performance Status, Fuhrman nuclear grading and TNM pathological stage, whereas the SSIGN score takes Stage, Size, Grade and Necrosis into consideration. Approximately 16% of patients with RCC present with Locoregional disease, and up to 40% of these patients relapse with metastatic disease, following nephrectomy. The 5-year survival for locoregional (stage III) disease is 53%, and 8% for metastatic disease. The standard management of high risk patients following nephrectomy has been surveillance, as there has been limited data demonstrating the benefit of adjuvant therapy in reducing the risk of relapse. Adjuvant therapy with immune check point inhibitors therapy is a potentially attractive treatment strategy for this patient group.
KEYNOTE-564 is a multicenter, double-blind, Phase III trial in which the benefit of adjuvant therapy with KEYTRUDA® was compared with placebo, following nephrectomy, in patients with clear cell RCC. In this study, 994 patients were randomized 1:1 to receive either KEYTRUDA® or placebo at least 12 weeks after surgery. Enrolled patients had histologically confirmed clear cell RCC, with Intermediate-High risk (pT2, Grade 4 or Sarcomatoid, N0 M0; or pT3, any Grade, N0 M0), High risk (pT4, any Grade, N0 M0; or pT any Stage, any Grade, N+ M0), or M1 with No Evidence of Disease after primary tumor and soft tissue metastases were completely resected, 1 year or less from nephrectomy. Treatment consisted of KEYTRUDA® 200 mg IV every 3 weeks (N=496) or placebo (N=498), every 3 weeks, for approximately 1 year. Both treatment groups were well balanced. The Primary end point of the trial was Disease Free Survival (DFS) assessment in all randomized patients and Secondary end points included Overall Survival (OS) and Safety. The median follow up at the time of data cut-off was 24.1 months.
At first prespecified interim analysis, the Primary endpoint of DFS was met. The median DFS was not reached for both treatment groups. KEYTRUDA® reduced the risk of recurrence or death by 32% compared with placebo, and this difference was statistically significant (HR=0.68; P=0.0010). The estimated DFS rate at 24 months was 77.3% with KEYTRUDA® versus 68.1% with placebo and this DFS benefit was consistent across subgroups. The estimated OS rate at 24 months was 96.6% with KEYTRUDA® versus 93.5% with placebo. Survival data are premature and additional follow up is planned for OS.
It was concluded that KEYTRUDA® demonstrated a statistically significant and clinically meaningful improvement in Disease Free Survival compared to placebo, in patients with Renal Cell Carcinoma, with a high risk of recurrence. The authors added that this is the first positive Phase III study with a checkpoint inhibitor, in adjuvant Renal Cell Carcinoma, and these practice changing results support KEYTRUDA® as a potential new standard of care for this patient group.
Pembrolizumab versus placebo as post-nephrectomy adjuvant therapy for patients with renal cell carcinoma: Randomized, double-blind, phase III KEYNOTE-564 study. Choueiri TK, Tomczak P, Park SH, et al. J Clin Oncol 2021; 39: (suppl 15; abstr LBA5) DOI: 10.1200/JCO.2021.39.15_suppl.LBA5