
SUMMARY: The American Cancer Society estimates that about 43,800 new cases of thyroid cancer will be diagnosed in the United States in 2022 and about 2,230 patients will die of the disease. Differentiated Thyroid Cancer (DTC) is the most common endocrine malignancy and includes Papillary, Follicular, and Hürthle-cell cancers, with Papillary thyroid cancers accounting for 80% of them. Majority of patients with DTC have clinical Stage I or Stage II disease, with a recurrence rate of less than 5% and cancer-related death rates even lower. Risk factors for recurrence include tumor size, multifocality, capsular or angioinvasion, degree of cervical lymph node involvement, existence of BRAF V600E Mutation, and thyroglobulin levels more than 0.5 ng/mL, after thyroidectomy.
Even though Radioiodine (iodine-131) therapy is not recommended for patients with a unifocal microcarcinoma (10 mm or less in diameter) following thyroidectomy, Radioiodine therapy is generally offered to a majority of patients with low-risk thyroid cancer, both to ablate residual normal thyroid tissue and to treat unresectable persistent disease. The benefits of this intervention however remain controversial. .
The authors conducted a prospective, multicenter, randomized, Phase III Essai Stimulation Ablation 2 (ESTIMABL2) trial involving patients with low-risk thyroid cancer, to assess the non-inferiority of observation versus postoperative Radioiodine therapy, following thyroidectomy. In this study, a total of 776 patients with low-risk Differentiated Thyroid Cancer who were undergoing thyroidectomy were randomly assigned 1:1 to receive ablation with postoperative Radioiodine therapy at a dose of 1.1 GBq (N=389) or no Radioiodine therapy (N=387). Enrolled patients had Differentiated Thyroid Carcinoma (Papillary, Follicular, or Oncocytic/Hürthle-cell cancer), with a multifocal pT1a tumor or a pT1b tumor. None of the patients had regional lymph node involvement, extrathyroidal extension or aggressive histologic subtypes (tall-cell, clear-cell, columnar-cell, and diffuse sclerosing variants of Papillary thyroid cancer, poorly differentiated). The mean patient age was 52 years, and 83% were women, 96% had papillary tumors 81% had pT1b N0 or Nx disease. All patients had normal results on postoperative neck ultrasonography. The follow-up protocol consisted of the measurement of thyroglobulin and thyroglobulin antibodies in all patients at 10 months and yearly thereafter. Ultrasonography of the neck was performed in all patients 10 months and 3 years after randomization. Disease-related events included residual or recurrent disease on neck ultrasonography and a serum thyroglobulin level of more than 1 ng/mL in the group receiving radioiodine and a level of more than 5 ng/mL in the nontreated group. No diagnostic Radioiodine scanning was performed after the whole-body scanning that was performed after therapy. The Primary objective was to assess whether no Radioiodine therapy was noninferior to Radioiodine therapy, with respect to the absence of a composite end point that included functional, structural, and biologic abnormalities, indicating residual or recurrent disease at 3 years.
After 3 years of follow up, there were no clinically meaningful differences in any of the end points between the two groups and the percentage of patients without an event was 95.6% in the no-Radioiodine therapy group and 95.9% in the Radioiodine therapy group, a result that met the noninferiority criteria. Events were more frequent in patients with a postoperative serum thyroglobulin level of more than 1 ng/mL during thyroid hormone treatment. BRAF V600E molecular alterations, which are associated aggressive tumor characteristics, were found in approximately 50% of the samples in each treatment group. The mutational status did not influence event rates in these low-risk patients. No treatment-related adverse events were reported and there was no difference in Quality-of-Life scores between the two groups.
It was concluded that in patients with low-risk thyroid cancer undergoing thyroidectomy, follow up without the use of Radioiodine therapy was noninferior to an ablation strategy with Radioiodine therapy, suggesting that patients with low-risk disease generally do well, regardless of whether they receive Radioiodine therapy.
Thyroidectomy without Radioiodine in Patients with Low-Risk Thyroid Cancer. Leboulleux S, Bournaud C, Chougnet CN, et al. N Engl J Med 2022; 386:923-932
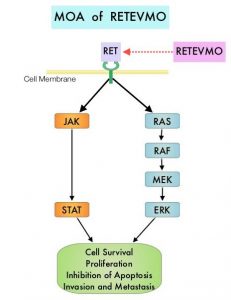
 The discovery of genetic alterations in the MAP Kinase pathway as well as the PI3K (Phosphatidylinositol-3-Kinase)-AKT-mTOR pathway in thyroid tumors, has lead to the development of Tyrosine Kinase Inhibitors (TKI’s), to target these activated pathways. LENVIMA® is an oral multitargeted TKI which targets Vascular Endothelial Growth Factor Receptor (VEGFR)1-3, Fibroblast Growth factor Receptor (FGFR)1-4, Rearranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with antiangiogenesis properties by its ability to inhibit FGFR-1 thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting by FGFR and PDGFR beta.
The discovery of genetic alterations in the MAP Kinase pathway as well as the PI3K (Phosphatidylinositol-3-Kinase)-AKT-mTOR pathway in thyroid tumors, has lead to the development of Tyrosine Kinase Inhibitors (TKI’s), to target these activated pathways. LENVIMA® is an oral multitargeted TKI which targets Vascular Endothelial Growth Factor Receptor (VEGFR)1-3, Fibroblast Growth factor Receptor (FGFR)1-4, Rearranged during Transfection tyrosine kinase receptor (RET), c-KIT, and Platelet Derived Growth Factor Receptor (PDGFR). LENVIMA® differs from other TKIs with antiangiogenesis properties by its ability to inhibit FGFR-1 thereby blocking the mechanisms of resistance to VEGF/VEGFR inhibitors. In addition, it controls tumor cell growth by inhibiting RET, c-KIT, and PDGFR beta and influences tumor microenvironment by inhibiting by FGFR and PDGFR beta.  The SELECT trial is a double-blind, multicenter, phase III study in which 392 patients with advanced RAI-refractory Differentiated Thyroid Cancer (DTC) were randomly assigned in a 2:1 ratio to receive LENVIMA® 24 mg PO daily in 28-day cycles (N=261) or placebo (N=131). Both treatment groups were well balanced and pretreatment with one prior Tyrosine Kinase Inhibitor (TKI) was allowed. Patients in the placebo group were allowed to cross over and receive open-label LENVIMA®, at the time of disease progression. The primary end point was Progression Free Survival (PFS) and secondary end points included Response Rate, Overall Survival, and safety. The median Progression Free Survival was 18.3 months in the LENVIMA® group and 3.6 months in the placebo group (HR= 0.21; P<0.001). This benefit in PFS associated with LENVIMA® was observed in all pre-specified subgroup of patients including those who had received one prior treatment with a TKI. The objective response rate with LENVIMA® was 64.8% versus 1.5% with placebo (P<0.001) and the median overall survival was not reached in either group. The most frequently reported grade 3 or more adverse events in the LENVIMA® group included hypertension (42.9%) and proteinuria (10%). Approximately 14% of the patients in the LENVIMA® group discontinued the drug due to adverse effects. Exploratory biomarker analyses were performed for BRAF and RAS mutations on tumor tissue and it was noted that LENVIMA® benefitted patients regardless of BRAFor RAS mutation status. The authors concluded that LENVIMA® decreased the risk of disease progression by 79% as compared with placebo and was associated with significant improvements in objective Response Rate among patients with RAI-refractory thyroid cancer. NEXAVAR® (Sorafenib), another multitargeted TKI, is presently available for this group of patients and therefore proper sequencing of LENVIMA® and NEXAVAR® remains unknown although it appears that LENVIMA® has a markedly higher Progression Free Survival compared to NEXAVAR®. Schlumberger M, Tahara M, Wirth LJ, et al. N Engl J Med 2015; 372:621-630
The SELECT trial is a double-blind, multicenter, phase III study in which 392 patients with advanced RAI-refractory Differentiated Thyroid Cancer (DTC) were randomly assigned in a 2:1 ratio to receive LENVIMA® 24 mg PO daily in 28-day cycles (N=261) or placebo (N=131). Both treatment groups were well balanced and pretreatment with one prior Tyrosine Kinase Inhibitor (TKI) was allowed. Patients in the placebo group were allowed to cross over and receive open-label LENVIMA®, at the time of disease progression. The primary end point was Progression Free Survival (PFS) and secondary end points included Response Rate, Overall Survival, and safety. The median Progression Free Survival was 18.3 months in the LENVIMA® group and 3.6 months in the placebo group (HR= 0.21; P<0.001). This benefit in PFS associated with LENVIMA® was observed in all pre-specified subgroup of patients including those who had received one prior treatment with a TKI. The objective response rate with LENVIMA® was 64.8% versus 1.5% with placebo (P<0.001) and the median overall survival was not reached in either group. The most frequently reported grade 3 or more adverse events in the LENVIMA® group included hypertension (42.9%) and proteinuria (10%). Approximately 14% of the patients in the LENVIMA® group discontinued the drug due to adverse effects. Exploratory biomarker analyses were performed for BRAF and RAS mutations on tumor tissue and it was noted that LENVIMA® benefitted patients regardless of BRAFor RAS mutation status. The authors concluded that LENVIMA® decreased the risk of disease progression by 79% as compared with placebo and was associated with significant improvements in objective Response Rate among patients with RAI-refractory thyroid cancer. NEXAVAR® (Sorafenib), another multitargeted TKI, is presently available for this group of patients and therefore proper sequencing of LENVIMA® and NEXAVAR® remains unknown although it appears that LENVIMA® has a markedly higher Progression Free Survival compared to NEXAVAR®. Schlumberger M, Tahara M, Wirth LJ, et al. N Engl J Med 2015; 372:621-630 The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this could impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4) The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this may impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)
The DECISION trial is a randomized, double-blind, multicenter phase III study in which the efficacy and safety of NEXAVAR® was compared with placebo, in patients with progressive RAI-refractory DTC. Four hundred and seventeen patients (417) were randomized to receive either NEXAVAR® 400 mg PO BID (n=207) or placebo (n=210). The median age was 63 yrs and only patients who had no prior chemotherapy or targeted therapy and with disease progression within the preceding 14 months, were included. Over 95% of the patients had metastatic disease and the most common sites of spread were lungs and lymph nodes. Treatment was continued until disease progression or until unacceptable toxicity was noted. Upon progression, patients in the placebo group were allowed to crossover and receive open-label NEXAVAR®. The primary endpoint was Progression Free Survival (PFS). Secondary endpoints included Overall Survival (OS), Response Rate (RR=Complete + Partial Response [PR]), and safety. The median PFS was 10.8 months with NEXAVAR® compared to 5.8 months with placebo (hazard ratio [HR] = 0.58; P <0.0001). Partial responses were observed in 12.2% of patients receiving NEXAVAR® compared with 0.5% in the placebo arm (P < 0.0001). The median duration of partial response was 10.2 months. Further, 42% of patients in the NEXAVAR® group had stable disease for 6 months or more compared to 33% in the placebo group. Median OS has not been reached. It should be noted that approximately 70% of patients in the placebo group were allowed to crossover to receive open-label NEXAVAR® and this may impact the OS data. The most common adverse events in the NEXAVAR® group included hand–foot skin reactions, diarrhea, rash/desquamation, fatigue and hypertension. The authors concluded that NEXAVAR® nearly doubled the PFS compared to placebo, in this select group of patients with advanced DTC and is the first and only FDA approved therapy for Differentiated Thyroid Cancers. Brose MS, Nutting C, Jarzab B, et al. J Clin Oncol 31, 2013 (suppl; abstr 4)