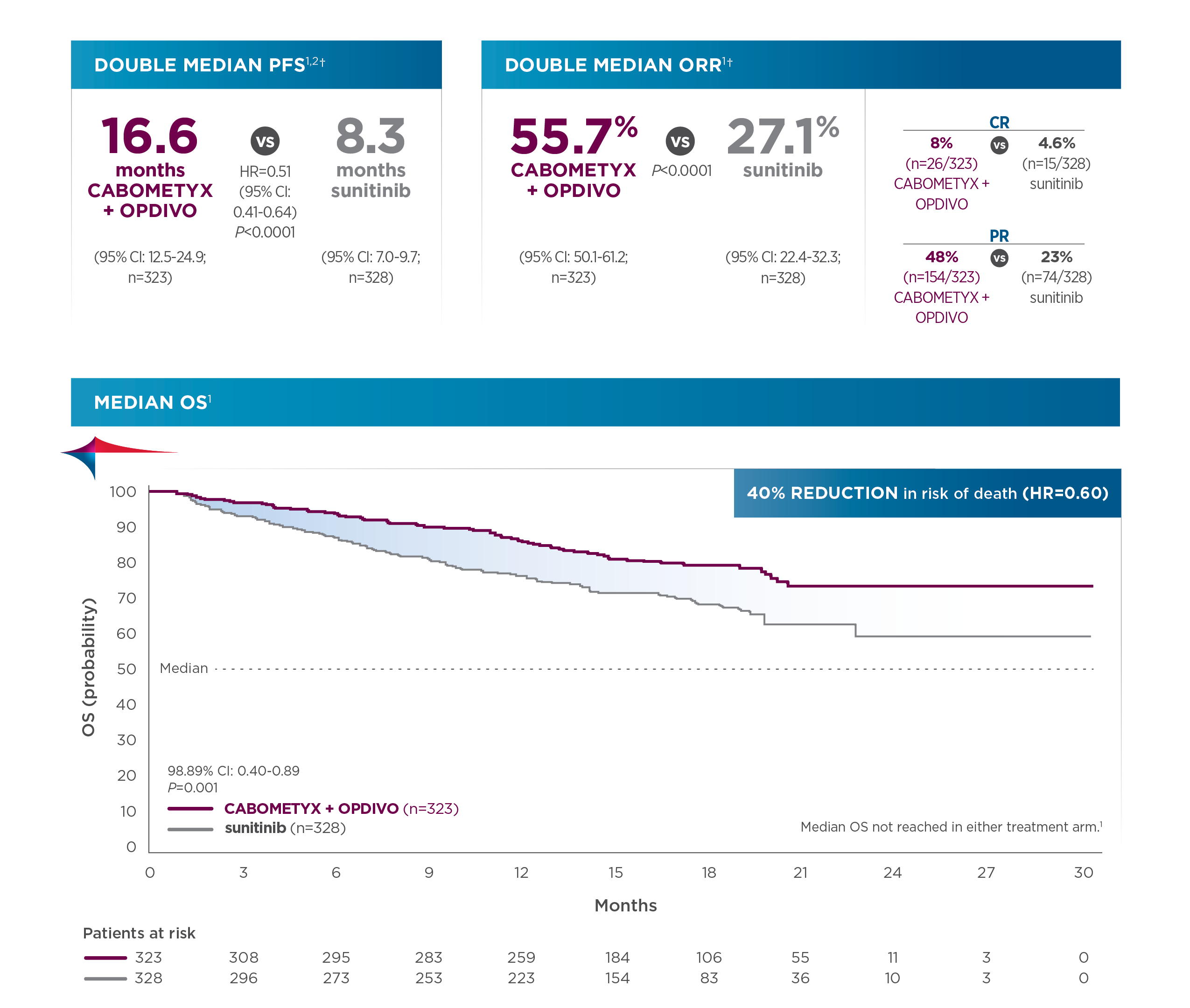
SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (12%) will develop invasive breast cancer during their lifetime. Approximately 290,560 new cases of breast cancer will be diagnosed in 2022 and about 43,780 individuals will die of the disease, largely due to metastatic recurrence.
Patients with early stage breast cancer often receive adjuvant therapy. Tumor biomarker assays have become an integral part of the treatment decision making process along with clinical and histologic tumor characteristics, further enabling customized care for patients with early-stage invasive breast cancer. A multitude of biomarker assays are presently available for the practicing Health Care Provider. Choosing the appropriate biomarker assay for a given patient can be a daunting task and the ASCO guidelines set forth herein, were developed by an expert panel based on a systematic review of evidence published from January 2016 to October 2021, of 24 Phase III randomized clinical trials (RCTs), prospective-retrospective studies, and clinical experience. These guidelines are only applicable for patients with newly diagnosed, non-metastatic, primary breast cancer, to prognosticate and predict outcomes but they do not however comment on the choice of specific treatment or regimens based on recurrence score. Treatment decisions should take into consideration disease stage, comorbidities and patient preferences. Even though several tests are now recommended in the guidelines, only one test should be used to guide therapy for an individual patient.
Three important questions were addressed by this guideline update
- For patients with early-stage ER-positive, HER2-negative breast cancer, which biomarkers should be used to guide decisions on adjuvant endocrine and chemotherapy for a newly diagnosed cancer or in the extended setting?
- For patients with early-stage HER2-positive breast cancer, which biomarkers should be used to guide decisions on adjuvant endocrine and chemotherapy?
- For patients with early-stage triple-negative breast cancer, which biomarkers should be used to guide decisions on adjuvant chemotherapy?
Newly Diagnosed ER-Positive, HER2-Negative Breast Cancer
Oncotype DX (21-gene recurrence score, 21-gene RS).
Recommendation 1.1. If a patient has node-negative breast cancer, the clinician may use the Oncotype DX test to guide decisions for adjuvant endocrine and chemotherapy
Recommendation 1.2. In the group of patients in Recommendation 1.1 with Oncotype DX recurrence score ≥ 26, the clinician should offer chemoendocrine therapy
Recommendation 1.3. In the group of patients in Recommendation 1.1 who are 50 years of age or younger with Oncotype DX recurrence score 16 to 25, the clinician may offer chemoendocrine therapy
Recommendation 1.4. If a patient is postmenopausal and has node-positive breast cancer with 1-3 positive nodes, the clinician may use the Oncotype DX test to guide decisions for adjuvant endocrine and chemotherapy
Recommendation 1.5. In the group of patients in Recommendation 1.4, the clinician should offer chemoendocrine therapy for those whose Oncotype DX recurrence score is ≥ 26
Recommendation 1.6. If a patient is premenopausal and has node-positive breast cancer with 1-3 positive nodes, the Oncotype DX test should not be offered to guide decisions for adjuvant systemic chemotherapy
Recommendation 1.7. If a patient has node-positive breast cancer with ≥ 4 positive nodes, the evidence on the clinical utility of routine Oncotype DX test to guide decisions for adjuvant endocrine and chemotherapy is insufficient to recommend its use
Qualifying statement: The genomic assay is prognostic and may be used for shared patient-physician treatment decision making.
MammaPrint (70-gene signature).
Recommendation 1.8. If a patient is older than 50 and has high clinical risk breast cancer that is node-negative or node-positive with 1-3 positive nodes, the clinician may use the MammaPrint test to guide decisions for adjuvant endocrine and chemotherapy
Recommendation 1.9. If a patient is 50 years of age or younger and has high clinical risk, node-negative or node-positive with 1-3 positive nodes breast cancer, the clinician should not use the MammaPrint test to guide decisions for adjuvant endocrine and chemotherapy
Recommendation 1.10. If a patient has low clinical risk, regardless of age, the evidence on clinical utility of routine MammaPrint test is insufficient to recommend its use
Recommendation 1.11. If a patient has node-positive breast cancer with ≥ 4 positive nodes, the evidence on the clinical utility of routine MammaPrint test to guide decisions for adjuvant endocrine and chemotherapy is insufficient to recommend its use
Qualifying statement: The genomic assay is prognostic and may be used for shared patient-physician treatment decision making.
EndoPredict (12-gene risk score).
Recommendation 1.12. If a patient is postmenopausal and has breast cancer that is node-negative or node-positive with 1-3 positive nodes, the clinician may use the EndoPredict test to guide decisions for adjuvant endocrine and chemotherapy
Recommendation 1.13. If a patient is premenopausal and has breast cancer that is node-negative or node-positive with 1-3 positive nodes, the clinician should not use the EndoPredict test to guide decisions for adjuvant endocrine and chemotherapy
Recommendation 1.14. If a patient has breast cancer with ≥ 4 positive nodes, evidence on the clinical utility of routine use of the EndoPredict test to guide decisions for adjuvant endocrine and chemotherapy is insufficient
Prosigna (PAM50).
Recommendation 1.15. If a patient is postmenopausal and has breast cancer that is node-negative, the clinician may use the Prosigna test to guide decisions for adjuvant systemic chemotherapy
Recommendation 1.16. If a patient is premenopausal and has node-negative or node-positive breast cancer, the clinician should not use the Prosigna test to guide decisions for adjuvant systemic chemotherapy
Recommendation 1.17. If a patient is postmenopausal and has node-positive breast cancer with 1-3 positive nodes, the evidence is inconclusive to recommend the use of the Prosigna test to guide decisions for adjuvant endocrine and chemotherapy
Recommendation 1.18. If a patient has node-positive breast cancer with ≥ 4 positive nodes, evidence on the clinical utility of routine use of the Prosigna test to guide decisions for adjuvant endocrine and chemotherapy is insufficient to recommend its use
Ki67.
Recommendation 1.19. If a patient is postmenopausal and has stage I-II breast cancer, the clinician may use Ki67 expression in conjunction with other clinical and pathologic parameters to guide decisions on adjuvant endocrine and chemotherapy when multigene assays are not available. Ki67 expression levels are most informative for prognosis when the level is < 5% (low proliferation) or > 30% (high proliferation) because technical reliability of distinguishing values within this range is limited
Recommendation 1.20. If a patient is postmenopausal and has breast cancer, there is insufficient evidence to use baseline Ki67 expression or Ki67 level after 2 weeks of neoadjuvant aromatase inhibitor (AI) therapy to guide decisions on adjuvant endocrine and chemotherapy
Recommendation 1.21. Despite the limitations associated with Ki67 testing, a patient with node-positive breast cancer with a high risk of recurrence and a Ki67 score of ≥ 20% as determined by a US Food and Drug Administration (FDA)–approved test may be offered 2 years of abemaciclib plus endocrine therapy
Immunohistochemistry 4.
Recommendation 1.22. If a patient has node-negative or node-positive breast cancer with 1-3 positive nodes, the clinician may use immunohistochemistry 4 (IHC4) score to guide decisions for adjuvant endocrine and chemotherapy if the score has been validated in the performing laboratory and if multigene assays are not available
Extended Endocrine Therapy for ER-Positive HER2-Negative Breast Cancer
Oncotype DX, EndoPredict, Prosigna, Ki67, or IHC4.
Recommendation 1.23. If a patient has node-negative breast cancer and has had 5 years of endocrine therapy without evidence of recurrence, there is insufficient evidence to use Oncotype DX, EndoPredict, Prosigna, Ki67, or IHC4 scores to guide decisions about extended endocrine therapy
Breast Cancer Index.
Recommendation 1.24. If a patient has node-negative or node-positive breast cancer with 1-3 positive nodes and has been treated with 5 years of primary endocrine therapy without evidence of recurrence, the clinician may offer the BCI test to guide decisions about extended endocrine therapy with either tamoxifen, an AI, or a sequence of tamoxifen followed by AI
Recommendation 1.25. If a patient has node-positive breast cancer with ≥ 4 positive nodes and has been treated with 5 years of primary endocrine therapy without evidence of recurrence, there is insufficient evidence to use the BCI test to guide decisions about extended endocrine therapy with either tamoxifen, an AI, or a sequence of tamoxifen followed by AI
Clinical treatment score post-5 years.
Recommendation 1.26. If a patient is postmenopausal and had invasive breast cancer and is recurrence-free after 5 years of adjuvant endocrine therapy, the clinical treatment score post-5 years (CTS5) web tool may be used to calculate the estimated risk of late recurrence (recurrence between years 5-10), which could assist in decisions about extended endocrine therapy
HER2-Positive Breast Cancer or Triple-Negative Breast Cancer
Oncotype DX, EndoPredict, MammaPrint, BCI, Prosigna, Ki67, or IHC4.
Recommendation 1.27. If a patient has HER2-positive breast cancer or TNBC, the clinician should not use multiparameter gene expression or protein assays (Oncotype DX, EndoPredict, MammaPrint, BCI, Prosigna, Ki67, or IHC4) to guide decisions for adjuvant endocrine and chemotherapy
Emerging Biomarkers
Tumor-infiltrating lymphocytes.
Recommendation 1.28. If a patient has node-negative or node-positive ER-positive, HER2-positive, or TNBC, the clinician should not use TILs to guide decisions for (neo)adjuvant endocrine and chemotherapy
PD-L1 testing.
Recommendation 1.29. If a patient has node-negative or node-positive ER-positive, HER2-positive, or TNBC, the clinician should not use PD-L1 testing to guide decisions for (neo)adjuvant endocrine and chemotherapy
Circulating tumor cells.
Recommendation 1.30. If a patient has node-negative or node-positive ER-positive, HER2-positive, or TNBC, the clinician should not use circulating tumor cells (CTC) to guide decisions for adjuvant endocrine and chemotherapy
Circulating tumor DNA.
Recommendation 1.31. If a patient has node-negative or node-positive ER-positive, HER2-positive, or TNBC, the clinician should not use ctDNA to guide decisions for adjuvant endocrine and chemotherapy
ASCO additionally recommended that all patients should be given the opportunity to participate in cancer clinical trials.
Biomarkers for Adjuvant Endocrine and Chemotherapy in Early-Stage Breast Cancer: ASCO Guideline Update. Andre F, Ismaila N, Allison KH, et al. DOI: 10.1200/JCO.22.00069 Journal of Clinical Oncology 40, no. 16 (June 01, 2022) 1816-1837.