


Written By: David M. Waterhouse, MD, MPH & Anita Koshy, MD
This promotional educational activity is brought to you by Janssen Biotech, Inc., and is not certified for continuing medical education.
Dr. Waterhouse is a paid consultant writing on behalf of Janssen Biotech, Inc., and must present this information in compliance with FDA requirements applicable to Janssen Biotech, Inc.
It is estimated that approximately 237,000 people in the US will be diagnosed with lung cancer in 2022. Despite advancements in standard-of-care treatments for lung cancer, this disease remains the leading cause of cancer death in both males and females.1 Nonetheless, the burgeoning number of targeted therapies for some types of lung cancer, particularly non-small cell lung cancer (NSCLC), have allowed for improvements in mortality and survival.2 As of 2022, there are ~20 targeted therapies for ~9 actionable driver mutations in stage IV NSCLC.3,4 In order to determine optimal targeted therapies, the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) recommend comprehensive biomarker testing, like next-generation sequencing (NGS), for all eligible patients at diagnosis of advanced NSCLC.5
Common EGFR Mutations (Exon 19 deletion and Exon 21 [L858R] mutations)
Epidermal growth factor receptor (EGFR) is a potent oncogene commonly altered in NSCLC, and EGFR driver mutations may be found in as many as 28% of metastatic NSCLC patients.6 Tyrosine kinase inhibitors (TKIs) directed against EGFR were among the first molecular targeted agents used for treatment of advanced NSCLC.7 Initial studies of EGFR TKIs showed that patient characteristics associated with EGFR mutations, such as non-smoking status, female gender, East Asian origin, and adenocarcinoma histology suggested a greater benefit from EGFR TKIs compared with first-line chemotherapy.8 Later studies identified gene mutations that could target the kinase domain of EGFR and predicted response to such inhibitors. The variable deletions of at least 3 amino acid residues in exon 19, as well as the single point mutation leucine-858 to arginine (L858R) in exon 21, are often referred to as “common” activating EGFR mutations and represent the vast majority (90%) of all observed EGFR kinase domain mutations in NSCLC.8 (Figure 1)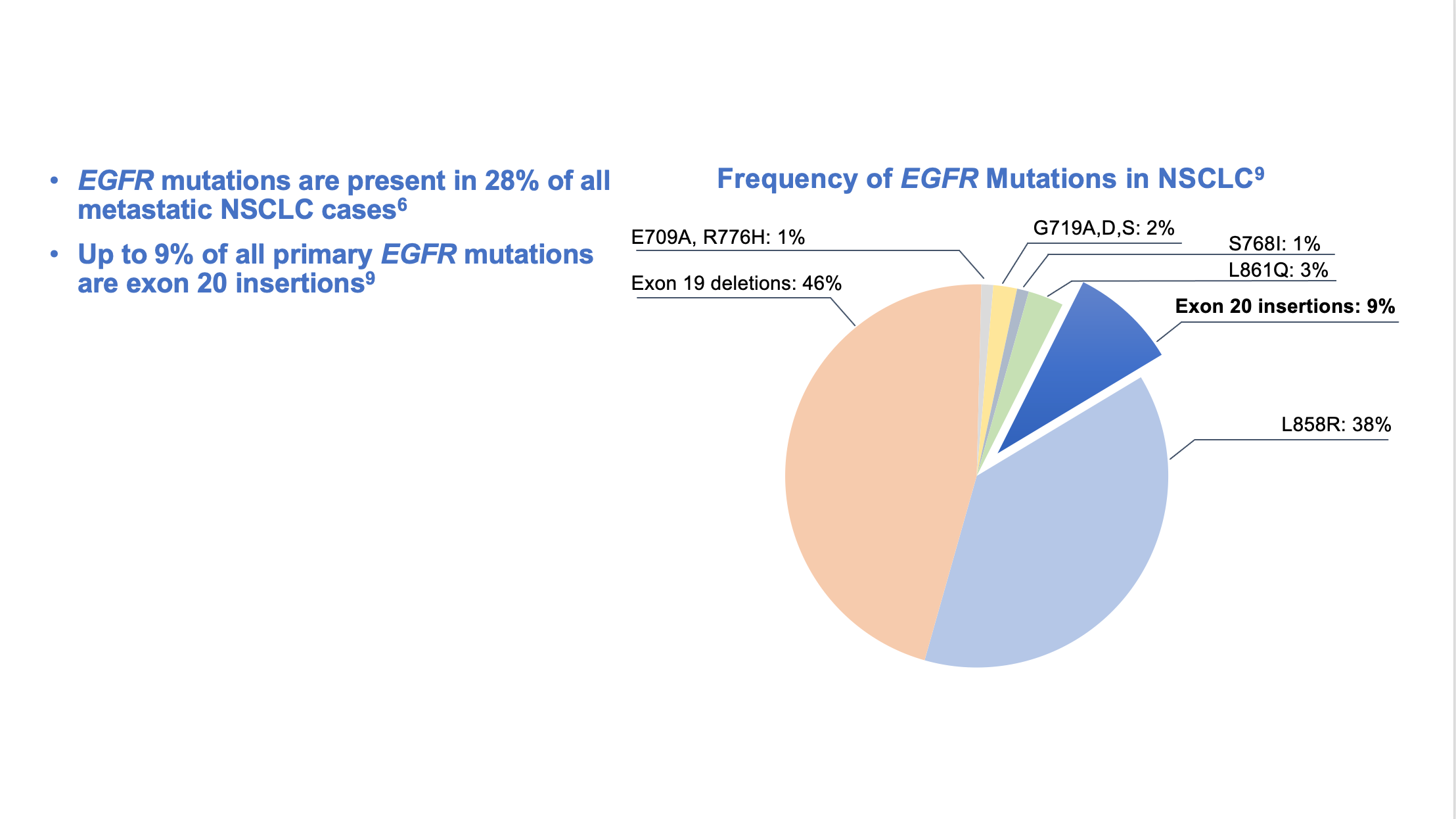
EGFR Exon 20 Insertion Mutations
Exon 20 insertion mutations are the third most prevalent type of activating EGFR mutations in NSCLC and are associated with a poor prognosis.9-11 These mutations are also enriched in women, non-smokers, Asian populations, and those with adenocarcinoma. Exon 20 insertion mutations, however, lack the key structural features that confer sensitivity of L858R and exon19 deletion mutations to first-and second-generation EGFR inhibitors. In-frame base pair insertions in exon 20 result in activation of EGFR, but, unlike the common activating EGFR mutations, they are associated with reduced affinity to most clinically available EGFR TKIs indicated for common EGFR mutations. Data are limited and variable, but multiple studies found that patients with EGFR exon 20 insertion mutations had an overall response rate of 0% to 8.7% when treated with first-, second-, or third-generation EGFR TKIs.12-16 (Figure 2)
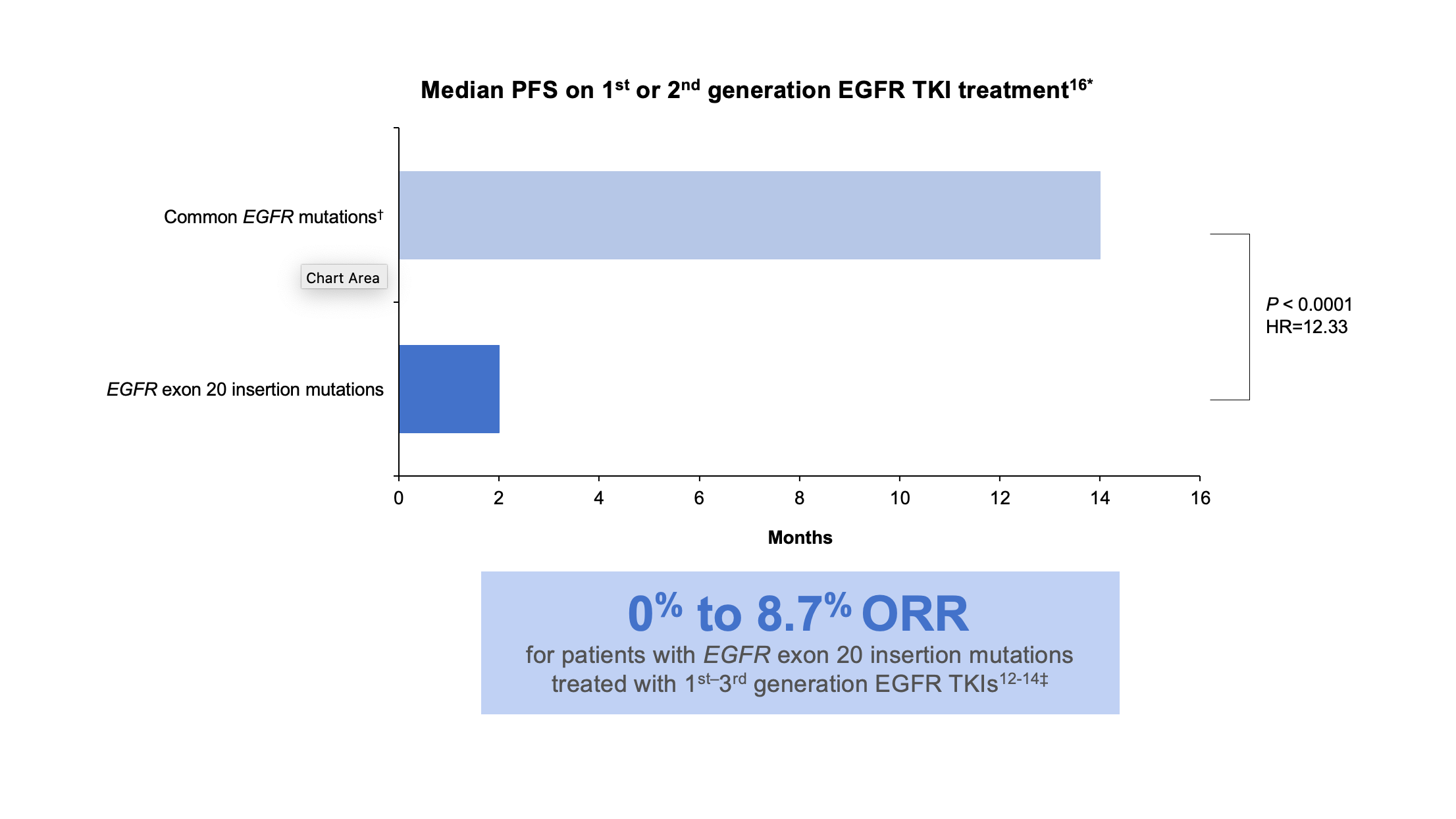
*These data were taken from a retrospective observational study.16
†Common mutations include L858R, L861Q, and exon 19 deletions.16
‡These data were taken from multiple sources: a cohort study, a prospective post hoc analysis of phase 2 and phase 3 trials, a single-center retrospective analysis, and a systematic literature review and meta-analysis.12-14
HR, hazard ratio; ORR, overall response rate; PFS, progression-free survival.
Study results also demonstrate limited efficacy of immuno-oncology (IO) monotherapy in this patient population compared to patients with wild-type EGFR. In a retrospective study using real-world data, patients with EGFR exon 20 insertion mutation-positive NSCLC were associated with a 58% increased risk of shorter time to next-line therapy after first-line IO monotherapy compared to patients with wild-type NSCLC.17
The NCCN Guidelines® do not recommend most TKIs or IO monotherapy for treating patients with mNSCLC and EGFR exon 20 insertion mutations in the first- or second-line setting. Instead, the Guidelines recommend platinum-based chemotherapy as the standard first-line treatment for NSCLC with EGFR exon 20 insertion mutations.5§
§Exceptions include p.A763_Y764insFQEA and p.A763_Y764insLQEA.5
EGFR Testing
The NCCN Guidelines recommend comprehensive biomarker testing, like NGS, prior to the initiation of first-line therapy, if clinically feasible.5 Despite that recommendation, rates of broad biomarker testing remain low, according to real-world evidence.18,19 In a retrospective observational chart review study among 3,474 patients with advanced NSCLC receiving first-line therapy in the US Oncology Network, the EGFR testing rate was found to be 70%, but comprehensive NGS testing was completed in only 42% of patients.20 Failure to order comprehensive NGS testing is particularly problematic when it comes to identifying EGFR exon 20 insertions. There are over 100 unique EGFR exon 20 insertion variants, and polymerase chain reaction (PCR) testing can miss approximately 50% of the insertions identified by NGS.21 (Figure 3)
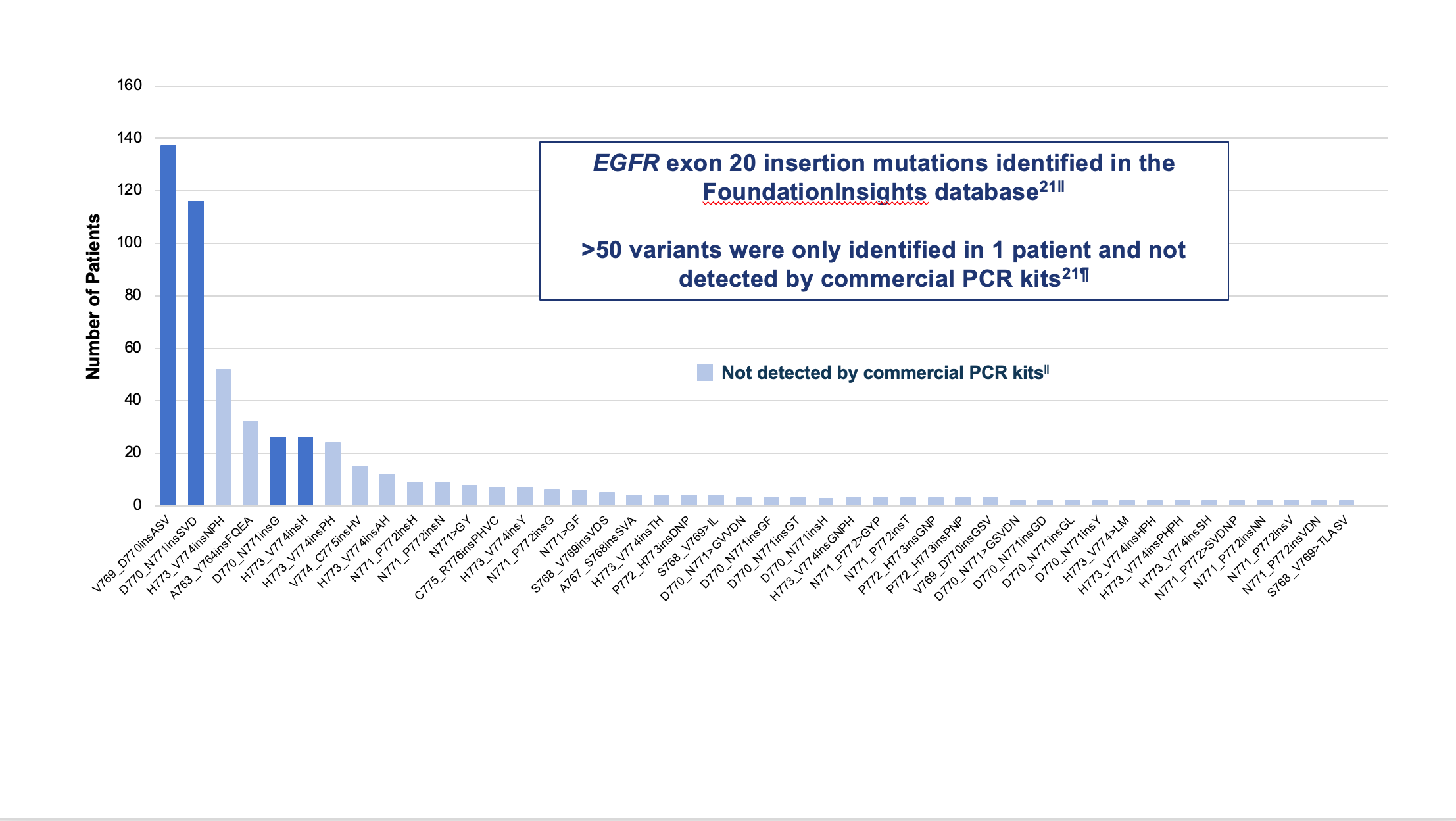
||Analysis from mutation profiles of 36,465 lung adenocarcinomas from Foundation Medicine (Cambridge, MA) FoundationInsights database, which is a database of 315,688 patient genomic profiles across 150 cancer types.
¶Commercially available qPCR methods were Roche cobas® EGFR mutation test v2 and Qiagen therascreen EGFR RGQ PCR kit.
Another notable issue is the accurate application of NGS data to clinical care. In multiple retrospective, observational cohort studies, approximately 17% to 24% of treatment-naive and 14% to 22% of second-line patients with EGFR exon 20 insertions received EGFR TKIs.11,17,22** Studies also found that approximately 7% to 40% of treatment-naive and 26% to 41% of second-line patients received IO monotherapy.17,22,23 These therapies (ie, most TKIs indicated for common mutations†† and IO monotherapies) are not recommended for first- or second-line therapy for EGFR exon 20 insertion mutations.5
**EGFR TKIs included first-, second- and third-generations.
††Exceptions include p.A763_Y764insFQEA and p.A763_Y764insLQEA.
Current Treatment Strategies for Patients With Exon 20 Insertion Mutations
Chemotherapy with a platinum doublet remains the recommended treatment option for the first-line treatment of patients with an EGFR exon 20 insertion mutation.5 When many of these patients progress, subsequent treatment options are needed. The NCCN Guidelines recommend amivantamab-vmjw or mobocertinib as subsequent therapy options for patients with EGFR exon 20 insertion mutations who have progressed on or after initial systemic therapy.5
Conclusion:
- Advances made in the treatment of NSCLC have improved patient mortality and survival,2 and these advancements are due in part to the discovery of actionable mutations, like common EGFR mutations, and targeted therapies3,4,7,8
- Multiple studies have found, however, that patients with EGFR exon 20 insertion mutations had a poor overall response when treated with first-, second-, or third-generation EGFR TKIs,11-15,17 and that IO monotherapies provide little benefit as a first-line treatment in patients with EGFR mutations, including exon 20 insertions17
- The NCCN Guidelines recommend:
- Testing eligible patients with mNSCLC for targetable genetic alterations to both identify potentially appropriate targeted therapies and avoid therapies unlikely to provide clinical benefit5
- Treating patients who harbor a common EGFR mutation (exon 19 deletion and exon 21 [L858R] mutations) with an EGFR TKI in the first line of treatment, whereas those with an EGFR exon 20 insertion mutation are best treated with a regimen containing a platinum doublet5
- Amivantamab-vmjw or mobocertinib as subsequent therapy options for patients with EGFR+ mNSCLC with exon 20 insertion mutations who have progressed on or after initial systemic therapy per the NCCN Guidelines5
References
1. National Cancer Institute. Cancer stat facts: common cancer sites. Accessed September 30, 2022. https://seer.cancer.gov/statfacts/html/common.html
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021.CA Cancer J Clin. 2021;71:7-33.
3. Benjamin DJ, Haslam A, Gill J, Prasad V. Targeted therapy in lung cancer: Are we closing the gap in years of life lost? Cancer Med. 2022;11(18):3417-3424.
4. Targeted Therapy in Metastatic Non–Small Cell Lung Cancer: Recent Updates and Controversies. Angel Qin. ASCO Daily News. Published January 19, 2022. Accessed November 14, 2022. https://dailynews.ascopubs.org/do/10.1200/ADN.22.200810/
5. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V.6.2022. © National Comprehensive Cancer Network, Inc. 2022. All rights reserved. Accessed December 2, 2022. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in anyway.
6. Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7(6):596-609.
7. Luo SY, Lam DC. Oncogenic driver mutations in lung cancer. Transl Respir Med. 2013;1(1):6.
8. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28 (Suppl 1):S24-S31.
9. Arcila ME, Nafa K, Chaft JE, et al. EGFR exon20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013;12(2):220-229.
10. Leal JL, Alexander M, Itchins M, et al. EGFR exon 20 insertion mutations: clinicopathological characteristics and treatment outcomes in advanced non-small cell lung cancer. Clin Lung Cancer. 2021;22(6):e859-e869.
11. Bazhenova L, Minchom A, Viteri S, et al. Comparative clinical outcomes for patients with advanced NSCLC harboring EGFR exon 20 insertion mutations and common EGFR mutations. Lung Cancer. 2021;162:154-161.
12. Wu JY, Yu CJ, Shih JY. Effectiveness of treatments for advanced non-small-cell lung cancer with exon 20 insertion epidermal growth factor receptor mutations. Clin Lung Cancer. 2019;20:e620-e630.
13. Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6.Lancet Oncol. 2015;16(7):830-838.
14. Kate S, Chougule A, JoshiA, et al. Outcome of uncommon EGFR mutation positive newly diagnosed advanced non-small cell lung cancer patients: a single center retrospective analysis. Lung Cancer (Auckl). 2019;10:1-10.
15. Kwon CS, Lin HM, Crossland V, et al. Non-small cell lung cancer with EGFR exon 20 insertion mutation: a systematic literature review and meta-analysis of patient outcomes. Curr Med Res Opin. 2022;38(8):1341-1350.
16. Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med. 2018;24:638-646.
17. Girard N, Minchom A, Ou SI, et al. Comparative clinical outcomes between EGFR ex20 ins and wild type NSCLC treated with immune checkpoint inhibitors. Clin Lung Cancer. 2022;23(7):571-577.
18. Paz-Ares L, Gondos A, Saldana D, et al. Genomic testing among patients with newly diagnosed advanced non-small cell lung cancer in the United States: A contemporary clinical practice patterns study. Lung Cancer. 2022;167:41-48.
19. Waterhouse DM, Tseng WY, Espirito JL, Robert NJ. Understanding contemporary molecular biomarker testing rates and trends for metastatic NSCLC among community oncologists. Clin Lung Cancer. 2021;22(6):e901-e910.
20. Robert N, Chen L, Espirito J, et al. Trends in molecular testing for metastatic non-small cell lung cancer in the US Oncology Network community practices. J Thorac Oncol. 2021;16(10) (suppl):S1169.
21. Bauml J, Viteri S, Minchom A, et al. Underdiagnosis of EGFR exon 20 insertion mutation variants: estimates from NGS-based real-world datasets. Presented at: the IASLC 2020 World Conference on Lung Cancer; January 28-31, 2021;Singapore.
22. He J, Pericone CD, Vanderpoel J. Real-world patient characteristics, treatment patterns, and mutation testing patterns among US patients with advanced non-small cell lung cancer harboring EGFR mutations. Adv Ther. 2022;39(7):3347-3360.
23. Choudhury NJ, Schoenfeld AJ, Flynn J, et al. Response to standard therapies and comprehensive genomic analysis for patients with lung adenocarcinoma with EGFR exon 20 insertions. Clin Cancer Res. 2021;27(10):2920-2927.
© Janssen Biotech, Inc. 2022 12/22 cp-345345v1