SUMMARY: The American Cancer Society estimates that in 2022, about 20,640 new cases of esophageal cancer will be diagnosed in the US and about 16,410 individuals will die of the disease. It is the sixth most common cause of global cancer death. Squamous Cell Carcinoma is the most common type of cancer of the esophagus among African Americans, while Adenocarcinoma is more common in Caucasians. Squamous Cell Carcinoma accounts for approximately 85% of cases.
Majority of esophageal cancers are unresectable at diagnosis, and most patients treated with curative intent eventually will relapse and only about 20% of patients will survive at least 5 years following diagnosis. Patients with advanced esophageal cancer have a median survival of less than a year when treated with the standard Fluoropyrimidine plus Platinum based chemotherapy. For those patients progressing on first line chemotherapy, treatment options are limited, with a 5-year relative survival rate of 8% or less.
OPDIVO® (Nivolumab) is a fully human, immunoglobulin G4 monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, thereby undoing PD-1 pathway-mediated inhibition of the immune response and unleashing the T cells. It has been noted that approximately 50% of patients with advanced esophageal Squamous Cell Carcinoma express tumor-cell Programmed Death Ligand 1 (PD-L1) greater than 1%. In the ATTRACTION-3 multicentre, Phase III trial, treatment with OPDIVO® was associated with a significant improvement in Overall Survival, compared with chemotherapy, in previously treated patients with advanced Esophageal Squamous Cell Carcinoma, regardless of PD-L1 expression. In the CheckMate 649 Phase III trial involving patients with gastric, gastroesophageal junction, or esophageal adenocarcinoma, first-line treatment with OPDIVO® plus chemotherapy resulted in a significant Overall Survival (OS) and Progression Free Survival (PFS) benefit, as compared with chemotherapy alone, as well as durable Objective Response Rate (ORR), with an acceptable safety profile.
CheckMate 648 is a global, open-label, Phase III trial in which the efficacy and safety of both an Immune Checkpoint Inhibitor in combination with chemotherapy and a dual Immune Checkpoint Inhibitor combination was evaluated in previously untreated patients with advanced esophageal Squamous Cell Carcinoma. The researchers herein reported the results for OPDIVO® plus chemotherapy and for OPDIVO® plus YERVOY® (Ipilimumab) as compared with chemotherapy alone.
In this study, 970 patients with previously untreated, unresectable, advanced, recurrent or metastatic esophageal Squamous Cell Carcinoma were randomly assigned 1:1:1 to receive OPDIVO® plus chemotherapy (N=321), OPDIVO® plus YERVOY® (N=325), or chemotherapy alone. Patients in the OPDIVO® plus chemotherapy group received OPDIVO® 240 mg IV every 2 weeks and chemotherapy consisted of Fluorouracil 800 mg/m2 IV Days 1-5 and Cisplatin 80 mg/m2 IV on Day 1, given every 4 weeks. The OPDIVO® plus YERVOY® group received OPDIVO® 3 mg/kg IV every 2 weeks plus YERVOY® 1 mg/kg IV every 6 weeks. Treatment was continued until disease progression or unacceptable toxicity. Patients could receive OPDIVO® or OPDIVO® plus YERVOY® for a maximum of 2 years. Demographic and baseline clinical characteristics were balanced across the treatment groups and in patients with tumor-cell PD-L1 expression of 1% or greater (49% of patients in each treatment group had tumor-cell PD-L1 expression of 1% or greater). The Primary end points were Overall Survival (OS) and Progression Free Survival (PFS), as determined by Blinded Independent Central Review (BICR), with hierarchical testing performed first in patients with tumor-cell PD-L1 expression of 1% or greater and then in the overall population. The Secondary end points included Objective Response Rate (ORR), which was also assessed by BICR.
After a minimum follow up period of 13 months, Overall Survival was significantly longer with OPDIVO® plus chemotherapy than with chemotherapy alone, both among patients with tumor-cell PD-L1 expression of 1% or greater (15.4 months versus 9.1 months; HR=0.54; P<0.001) and in the overall population (13.2 months versus 10.7 months; HR=0.74; P=0.002). These findings suggested a 46% and 26% lower risk of death respectively with OPDIVO® plus chemotherapy, than with chemotherapy alone. Overall Survival was also significantly longer with OPDIVO® plus YERVOY® than with chemotherapy among patients with tumor-cell PD-L1 expression of 1% or greater (13.7 months versus 9.1 months; HR=0.64; P=0.001) and in the overall population (12.7 months versus 10.7 months; HR=0.78; P=0.01).
There was a significant improvement in Progression Free Survival seen with OPDIVO® plus chemotherapy over chemotherapy alone among patients with tumor-cell PD-L1 expression of 1% or greater (HR=0.65; P=0.002). This PFS benefit was not seen with OPDIVO® plus YERVOY®, as compared with chemotherapy. The incidence of Grade 3 or 4 treatment-related Adverse Events was 47% with OPDIVO® plus chemotherapy, 32% with OPDIVO® plus YERVOY® and 36% with chemotherapy alone.
Treatment with either OPDIVO®-based regimens resulted in a higher Complete Response rate, as well as in more durable responses, than chemotherapy alone. Of the three treatment regimens, OPDIVO® plus chemotherapy led to the highest Objective Response Rate and OPDIVO® plus YERVOY® resulted in the longest median Duration of Response.
It was concluded that first-line treatment of advanced esophageal squamous-cell carcinoma with either OPDIVO® plus chemotherapy or OPDIVO® plus YERVOY® resulted in a significantly longer Overall Survival benefit and durable responses, than chemotherapy alone.
Nivolumab Combination Therapy in Advanced Esophageal Squamous-Cell Carcinoma. Doki Y, Ajani JA, Kato K, et al. N Engl J Med 2022;386:449-462

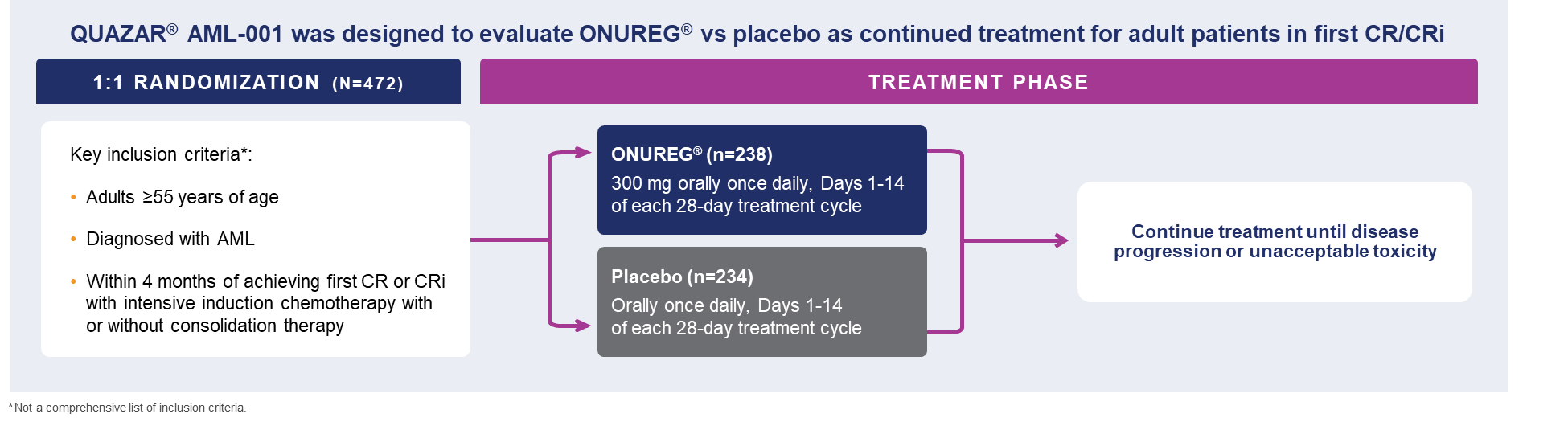 A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8
A total of 472 patients were randomized 1:1 to receive either ONUREG® 300 mg or placebo orally on Days 1 through 14 of each 28-day cycle.8 Baseline demographics and disease-related characteristics were well balanced between the ONUREG and placebo arms.8 Across both arms, 72% of patients were 65 years or older, and most patients (92%) had an ECOG PS of 0 or 1. Additionally, approximately three-quarters of patients received 1 or 2 cycles of consolidation therapy.8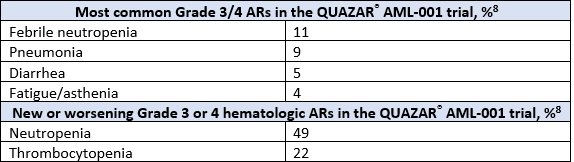
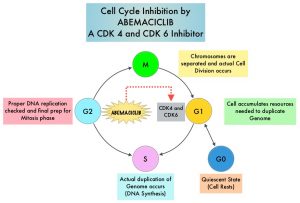 VERZENIO® (Abemaciclib) is an oral, selective inhibitor of CDK4 and CDK6 kinase activity, and prevents the phosphorylation and subsequent inactivation of the Rb tumor suppressor protein, thereby inducing G1 cell cycle arrest and inhibition of cell proliferation.
VERZENIO® (Abemaciclib) is an oral, selective inhibitor of CDK4 and CDK6 kinase activity, and prevents the phosphorylation and subsequent inactivation of the Rb tumor suppressor protein, thereby inducing G1 cell cycle arrest and inhibition of cell proliferation.