SUMMARY: The FDA on May 28, 2021, granted accelerated approval to LUMAKRAS® (Sotorasib), a RAS GTPase family inhibitor, for adult patients with KRAS G12C mutated locally advanced or metastatic Non Small Cell Lung Cancer (NSCLC), as determined by an FDA approved test, who have received at least one prior systemic therapy. The FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for LUMAKRAS®. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
The American Cancer Society estimates that for 2021, about 235,760 new cases of lung cancer will be diagnosed and 131,880 patients will die of the disease. Lung cancer is the leading cause of cancer-related mortality in the United States. Non-Small Cell Lung Cancer (NSCLC) accounts for approximately 85% of all lung cancers. Of the three main subtypes of NSCLC, 30% are Squamous Cell Carcinomas (SCC), 40% are Adenocarcinomas and 10% are Large Cell Carcinomas. With changes in the cigarette composition and decline in tobacco consumption over the past several decades, Adenocarcinoma now is the most frequent histologic subtype of lung cancer.
The KRAS (kirsten rat sarcoma viral oncogene homologue) proto-oncogene encodes a protein that is a member of the small GTPase super family. The KRAS gene provides instructions for making the KRAS protein, which is a part of a signaling pathway known as the RAS/MAPK pathway. By relaying signals from outside the cell to the cell nucleus, the protein instructs the cell to grow, divide and differentiate. The KRAS protein is a GTPase, and converts GTP into GDP. To transmit signals, the KRAS protein must be turned on, by binding to a molecule of GTP. When GTP is converted to GDP, the KRAS protein is turned off or inactivated, and when the KRAS protein is bound to GDP, it does not relay signals to the cell nucleus. The KRAS gene is in the Ras family of oncogenes, which also includes two other genes, HRAS and NRAS. When mutated, oncogenes have the potential to change normal cells cancerous.
KRAS is the most frequently mutated oncogene in human cancers and are often associated with resistance to targeted therapies and poor outcomes. The KRAS-G12C mutation occurs in approximately 12-15% of Non Small Cell Lung Cancers (NSCLC) and in 3-5% of Colorectal cancers and other solid cancers. KRAS G12C is one of the most prevalent driver mutations in NSCLC and accounts for a greater number of patients than those with ALK, ROS1, RET, and TRK 1/2/3 mutations combined. KRAS G12C cancers are genomically more heterogeneous and occur more frequently in current or former smokers, and are likely to be more complex genomically than EGFR mutant or ALK rearranged cancers. G12C is a single point mutation with a Glycine-to-Cysteine substitution at codon 12. This substitution favors the activated state of KRAS, resulting in a predominantly GTP-bound KRAS oncoprotein, amplifying signaling pathways that lead to oncogenesis.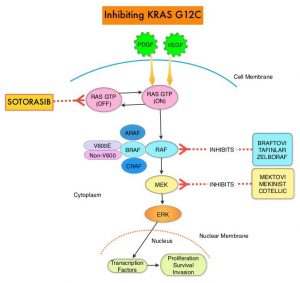
LUMAKRAS® is a first-in-class small molecule that specifically and irreversibly inhibits KRAS-G12C and traps KRAS-G12C in the inactive GDP-bound state. Preclinical studies in animal models showed that LUMAKRAS® inhibited nearly all detectable phosphorylation of Extracellular signal-Regulated Kinase (ERK), a key downstream effector of KRAS, leading to durable complete regression of KRAS-G12C tumors.
The CodeBreaK clinical development program for LUMAKRAS® was designed to treat patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers. This program has enrolled more than 800 patients across 13 tumor types since its inception.
CodeBreaK 100 is a Phase I and II, first-in-human, open-label, single arm, multicenter study, which enrolled patients with KRAS G12C-mutant solid tumors. Eligible patients must have received a prior line of systemic anticancer therapy, for their tumor type and stage of disease. The present FDA approval was based on a Phase II trial which enrolled 126 patients with NSCLC, 124 of whom had centrally evaluable lesions by RECIST criteria at baseline. Enrolled patients had locally advanced or metastatic NSCLC with a KRAS G12C mutation, who had progressed on an immune checkpoint inhibitor and/or platinum-based chemotherapy, and those with active brain metastases were excluded. Patient received LUMAKRAS® 960mg orally once daily, until disease progression or unacceptable toxicity. Imaging studies were done every 6 weeks up to week 48 and then once every 12 weeks thereafter. The Primary end point of the trial was Overall Response Rate (ORR) as assessed by blinded Independent Central Review. Secondary end points included Duration of Response (DOR), Disease Control Rate (DCR), time to recovery, Progression Free Survival (PFS), Overall Survival, and Safety. The examination of biomarkers served as an exploratory end point. Patients were followed for a median of 12.2 months.
The ORR was 37.1% and the median Duration of Response was 10 months. Three patients had a Complete Response and the Disease Control Rate was 80.6%. The median Time to response was 1.4 months and 72% of patients had an early rapid response on first CT scan at 6 weeks. Approximately 81% of patients had tumor shrinkage of any magnitude, and the median percentage of best tumor shrinkage among all responders was 60%, and these responses were durable. The median PFS was 6.8 months. In the exploratory biomarker analysis, tumor response to LUMAKRAS® was seen across subgroups, including patients with negative or low expression of PD-L1 and those with STK11 and TP53 mutations. The most common adverse reactions were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities were increase in liver function tests, anemia, hyponatremia and proteinuria.
It was concluded that patients with NSCLC have poor outcomes and limited treatment options following progression on first line treatment. LUMAKRAS® offers a new treatment option for this patient group, and it is the first KRAS-targeted therapy to be approved after nearly four decades of research. A global Phase III study (CodeBreaK 200) is underway, comparing LUMAKRAS® to Docetaxel in patients with KRAS G12C-mutated NSCLC.
CodeBreaK 100: Registrational Phase 2 Trial of Sotorasib in KRAS p.G12C Mutated Non-small Cell Lung Cancer. Li BT, Skoulidis F, Falchook G, et al. Presented at: International Association for the Study of Lung Cancer 2020 World Conference on Lung Cancer; January 28-31, 2021; virtual. Abstract PS01.07.