SUMMARY: Hereditary factors play an important role in the risk of developing several cancers. Therefore, identification of a germline predisposition can have important implications for treatment decision making, risk-reducing interventions, cancer screening for early diagnosis, and germline testing of unaffected relatives. Previously published studies have been biased by estimating the prevalence of germline cancer susceptibility in patients with breast, prostate, and colorectal cancer from registry populations, genetic testing companies, and high-risk cancer clinics. Very few studies have compared the prevalence of germline findings in patients with cancer, not selected by practice guidelines, and the impact of universal testing strategy for inherited germline variants in patients with cancer has remained unclear. The purpose of this present study was to determine if universal genetic testing in patients with cancer identifies more inherited cancer predisposition variants than a guideline-based approach, and also find out if there is an association between universal genetic testing and clinical management.
The authors in this prospective, multicenter cohort study, assessed germline genetic alterations among patients with solid tumors, receiving care at Mayo Clinic Cancer Centers and Mayo Clinic Health System community oncology practice in the US, between April 2018, and March 2020, as a part of 2 year Interrogating Cancer Etiology Using Proactive Genetic Testing (INTERCEPT) program. Patients were NOT selected based on cancer type, stage of disease, family history of cancer, race/ethnicity, age at diagnosis, multifocal tumors, or personal history of multiple malignant neoplasms. Clinical, demographic, and family history data and pathologic information were collected on all patients from medical records or self-administered questionnaires.
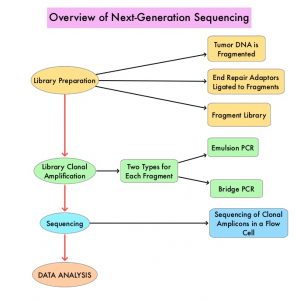
Germline sequencing using a Next-Generation Sequencing panel of 84 genes was offered at no cost, utilizing the Invitae Multi-Cancer Panel. Whole Genome Sequencing, deletion and duplication analysis, and variant interpretation were performed and Pathogenic Germline Variants (PGV) were classified as High (relative risk more than 4), Intermediate (relative risk, 2-4), or Low (relative risk less than 2) penetrance, or recessive medically actionable variants. Test results were disclosed to the patient, and those with Pathogenic Germline Variants (PGVs) were invited for genetic counseling.
The authors compared multi-gene panel testing with guideline-based testing, using guidelines from the National Comprehensive Cancer Network (NCCN) and the National Society of Genetic Counselors (NSGC) and the American College of Medical Genetics (ACMG), to determine whether genetic testing was indicated for a particular patient. For patients who met the guidelines, the only genes tested were those recommended by the tumor-specific guideline. This study included patients with a broad mix of cancer types at various stages. The final analytic cohort consisted of 2984 patients, out of the 3095 patients enrolled in the study. The mean patient age was 61 years, 53% were male and 44% of patients had Stage IV disease at the time of genomic analysis. A family history of cancer in a first-degree relative was reported in 34% of the participants. The goals of this study were to examine the proportion of Pathogenic Germline Variants (PGVs) detected with a universal testing strategy compared with a targeted testing strategy based on clinical guidelines, as well as uptake of cascade genetic testing in families, when offered at no cost.
It was noted that Pathogenic Germline Variants (inherited mutation in a gene) associated with the development of their cancer was found in 13.3% of patients, including moderate and high-penetrance cancer susceptibility genes. In this study, 1 in 8 patients had a PGV, half of which would not have been detected using a guideline-based testing approach. Of those with a high-penetrance PGVs, 28.2% had modifications in their treatment, based on the finding. About 6.4% had incremental clinically actionable findings that would not have been detected by phenotype or family history-based testing criteria. However, only 17.6% of participants with PGVs had family members undergoing no-cost cascade genetic testing when offered.
It was concluded that in this large, prospective, multicenter cohort study with a broad mixture of cancer types and stages, universal multigene panel testing was associated with increased detection of clinically actionable heritable variants, compared with a targeted testing strategy based on clinical guidelines. Approximately 30% of patients with high-penetrance variants had modifications in their treatment, suggesting that wider clinical implementation of universal genetic testing and acceptance in oncology practice, may be beneficial.
Comparison of Universal Genetic Testing vs Guideline-Directed Targeted Testing for Patients With Hereditary Cancer Syndrome. Samadder NJ, Riegert-Johnson D, Boardman L, et al. JAMA Oncol. Published online October 30, 2020. doi:10.1001/jamaoncol.2020.6252
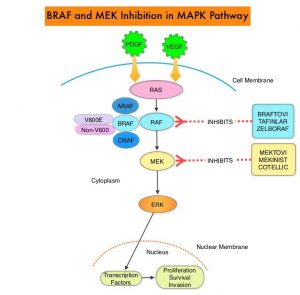
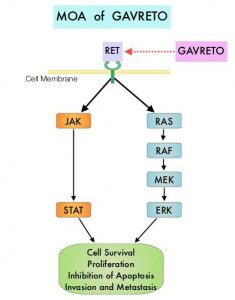 and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease.
and accounts for about 14% of all new cancers and 27% of all cancer deaths. The American Cancer Society estimates that for 2020, about 228, 820 new cases of lung cancer will be diagnosed and 135,720 patients will die of the disease.