SUMMARY: Breast cancer is the most common cancer among women in the US and about 1 in 8 women (13%) will develop invasive breast cancer during their lifetime. Approximately 276,480 new cases of invasive female breast cancer will be diagnosed in 2020 and about 42,170 women will die of the disease. Approximately 6% of newly diagnosed breast cancer patients present with Stage IV disease and about half of patients with primary breast cancer will progress later to the metastatic stage. About 70% of breast tumors express Estrogen Receptors and/or Progesterone Receptors and Hormone Receptor (HR)-positive/HER2-negative breast cancer is the most frequently diagnosed molecular subtype. Most of these patients with advanced disease in the current era are treated with a combination of CDK4/6 inhibitor and endocrine therapy (often an oral Aromatase Inhibitor), based on survival data. However, resistance to these regimens typically develops in a majority of the patients.
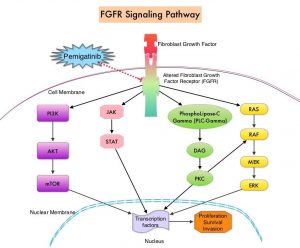
The PhosphoInositide 3-Kinase (PI3K) pathway is an intracellular signaling pathway important in the regulation of cancer cell proliferation and metastasis. PI3K is a lipid kinase and has four distinct isoforms – alpha, beta, gamma and delta, which play a unique role in the survival of different tumor types and establishment of supportive tumor microenvironments. The alpha and beta isoforms are expressed in a wide variety of tissues whereas the gamma and delta isoforms are primarily expressed in hematopoietic cells such as B and T cells. The PI3K alpha isoform is particularly important in breast cancer and plays an important role in tumorigenesis, supporting tumor angiogenesis and stromal interactions, making this a viable target. PIK3CA is an oncogene that codes for the alpha isoform of PI3K, (PI3Kα), more specifically for the alpha isoform of p110. The PI3k pathway is the most frequently altered pathway in human cancers including breast cancer, and has been implicated in disease progression in a significant number of patients with breast cancer. Activation of the PI3K pathway in breast cancer has been associated with resistance to endocrine therapy and disease progression. Approximately 40% of patients with Hormone Receptor positive (HR+), HER2-negative breast cancers, harbor activating mutations in the PIK3CA isoform of PI3K, which is the most common mutation in HR+ breast cancer. Patients with advanced breast cancer harboring PIK3CA mutations typically have a poor prognosis. This provides a strong rationale for targeting the PI3K pathway in breast cancer.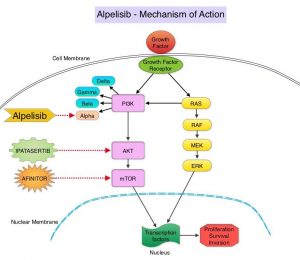
PIQRAY® is an oral, alpha-specific PI3K inhibitor that specifically inhibits PIK3 in the PI3K/AKT kinase signaling pathway. Further, it was shown in preclinical studies that cancer cells with PIK3CA mutations are more sensitive to PIQRAY® than those without the mutation, across a broad range of tumor types. In the SOLAR-1 Phase III trial, there was a 35% improvement in Progression Free Survival (PFS) in patients randomized to PIQRAY® plus FASLODEX®, compared to the placebo plus FASLODEX® group, among postmenopausal patients with PIK3CA-mutated, HR+/HER2- negative, advanced breast cancer, who had progressed on or following prior Aromatase Inhibitor (AI) treatment with or without a CDK 4/6 inhibitor. However in this study, only 6% had received prior CDK4/6 inhibitor therapy and there is presently limited data available, to inform treatment decisions in patients who progress on AI and CDK 4/6 inhibitor combination.
BYLieve is an ongoing, prospective, open-label, Phase II, non-comparative trial, which evaluated the benefit of PIQRAY® in combination with endocrine therapy in patients with HR+, HER-negative, PIK3CA-mutated, advanced breast cancer, who progressed on or after a prior therapy including CDK inhibitor. This study included 3 patient cohorts– Cohort A included patients who received a CDK4/6 inhibitor plus an AI as immediate prior therapy, Cohort B included patients who received a CDK4/6 inhibitor plus FASLODEX® (Fulvestrant) as immediate prior therapy, and Cohort C included patients who progressed on/after an AI and received chemotherapy or endocrine therapy as immediate prior treatment.
The authors in this publication shared findings from Cohort A group of patients, who had received CDK4/6 inhibitor plus an AI as their immediate prior therapy. Cohort A enrolled 127 patients of whom 121 patients had centrally confirmed PIK3CA mutation. Patients in Cohort A received PIQRAY® 300 mg orally once daily along with FASLODEX® 500 mg IM on Day 1 and 15 of cycle 1 followed by Day 1 treatment, of each 28 day cycle thereafter. The median patient age was 58 years. Seventy percent (70%) of patients had received one prior metastatic regimen, none of the patients had received FASLODEX® as a first-line metastatic agent, and 60% of patients had secondary endocrine resistance. The median follow up was 11.7 months. The Primary endpoint was proportion of patients alive without disease progression at 6 months. Secondary end points included Progression Free Survival (PFS), Overall Response Rate (ORR), Overall Survival (OS), and safety.
The Primary endpoint was met and the proportion of patients with confirmed PIK3CA mutation and without disease progression at 6 months was 50.4%. The median PFS was 7.3 months. Among the 121 patients in Cohort A with a confirmed PIK3CA mutation, the response rate, which was all partial responses was 17.4%, and 45.5% achieved stable disease.
Although the BYLieve trial did not have a control group to allow comparing patients in Cohort A to patients receiving other standard therapies, the authors conducted a weighted/matched analysis between the patients in Cohort A of the BYLieve trial and a Real-World similar group of 95 patients with HR+, HER2-negative, PIK3CA-mutated advanced breast cancer, who were treated with standard therapies. The Real-World patient data was obtained from the de-identified clinic-genomic database of Flatiron Health and Foundation Medicine. These 95 patients had received a wide range of regimens, with the most frequent being XELODA® (Capecitabine) monotherapy, FASLODEX® monotherapy, FASLODEX® plus IBRANCE® (Palbociclib), AFINITOR® (Everolimus) plus AROMASIN® (Exemestane), FASLODEX® plus FEMARA® (Letrozole), and IBRANCE® monotherapy.
Unadjusted results showed a median PFS of 7.3 months in BYLieve Cohort A versus 3.6 months in the Real-World cohort. Similar outcomes were noted when data were weighted by odds, propensity score matching, and exact matching.
It was concluded that the BYLieve trial is continuing to show clinically meaningful efficacy with a combination of PIQRAY® and FASLODEX® in HR+, HER2-negative, PIK3CA-mutated advanced breast cancer, post CDK inhibitor treatment, building further on the findings of SOLAR-1 trial. The matched analysis comparing BYLieve with Real-World Data in the post-CDK4/6 inhibitor setting, further supports use of PIQRAY® plus FASLODEX® for this patient group.
Alpelisib (ALP) + fulvestrant (FUL) in patients (pts) with PIK3CA-mutated (mut) hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2–) advanced breast cancer (ABC) previously treated with cyclin-dependent kinase 4/6 inhibitor (CDKi) + aromatase inhibitor (AI): BYLieve study results. Rugo HS, Lerebours F, Ciruelos E, et al. J Clin Oncol 38: 2020 (suppl; abstr 1006).