Author: Cristina Gasparetto, MD
Sponsored by: Karyopharm Therapeutics, Inc.
Dr. Gasparetto is a paid consultant for Karyopharm Therapeutics, Inc. and has been compensated.
Multiple myeloma (MM) remains an incurable hematologic cancer due to the clonal nature of the disease.1 With each relapse, cancer cells undergo clonal evolution and acquire new mutations that render them resistant to certain treatments.1 Triplet therapies combining proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), and anti-CD38 monoclonal antibodies (CD38-mAbs) have improved patient outcomes and their use has steadily increased over the past decade.2,3 When patients relapse after exposure to daratumumab (a CD38-mAb), the prognosis becomes unfavorable; even if patients previously responded to PIs or IMiDs, median survival may not reach one year.3 A significant unmet need therefore remains for providing durable disease control for patients with MM.1
For patients with previously-treated MM, the National Comprehensive Cancer Network® (NCCN®) recommends a new triplet regimen should preferably include drugs or drug classes patients have not been exposed to, or not exposed to for at least 6 months.4 For patients with MM who are triple class exposed, a selective inhibitor of nuclear export (SINE) may be a potential treatment class to consider in early relapsed (1-3 prior therapies) MM.4 Once-weekly XPOVIO® (selinexor), is a first-in-class, oral SINE compound approved as early as first relapse in MM that reversibly inhibits exportin 1 (XPO1).5 This action leads to accumulation of tumor suppressor proteins in the nucleus and reductions in several oncoproteins, such as c-myc and cyclin D1, cell cycle arrest, and apoptosis of cancer cells.5 Oral, once weekly selinexor (XPOVIO®) in combination with bortezomib and dexamethasone (XVd) is recommended by the NCCN as a Category 1 therapeutic option in early relapsed (1 to 3 prior therapies) MM.4
The efficacy and safety of XPOVIO was assessed in a phase 3, randomized, open-label trial comparing XPOVIO (100 mg once weekly) in combination with bortezomib (1.3 mg/m2) and dexamethasone (20 mg) with Vd alone in patients exposed to one to three prior lines of therapy.6 Patient disease characteristics were well balanced in both treatment groups and the primary endpoint was progression-free survival (PFS).5,6 Patients in the XVd group demonstrated a median PFS of 13.9 months (95% CI: 11.73-NE) compared with 9.5 months (95% CI: 8.11-10.78) in the Vd group (HR 0.70 [95% CI: 0.53-0.93], P=0.0075).6 In patients treated with XVd, a greater median PFS was consistently observed in certain subgroups compared with patients treated with Vd (Figure 1).6,7 When comparing patients 65 years of age and older to younger patients, older patients had a higher incidence of discontinuation due to an adverse reaction (28% vs 13%) and a higher incidence of serious adverse reactions (56% vs 47%).5
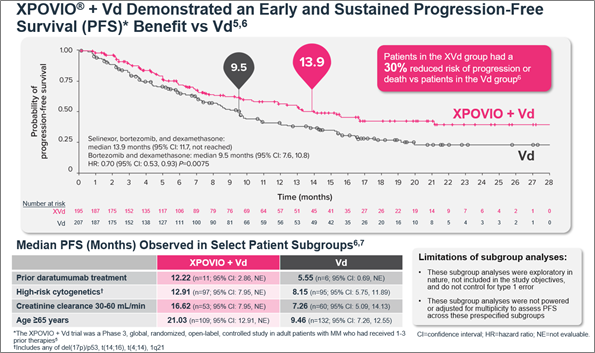 Figure 1. Median PFS in the XVd and Vd treatment groups (primary endpoint) and in select patient subgroups in the XVd trial.
Figure 1. Median PFS in the XVd and Vd treatment groups (primary endpoint) and in select patient subgroups in the XVd trial.
Oral, once-weekly XPOVIO dosage may be adjusted to help mitigate potential adverse reactions (ARs).5 The indicated starting dose of XPOVIO is 100 mg once weekly and the dose may be reduced to 80 mg, 60 mg, or 40 mg based on ARs.5 Dose reductions were permitted in the XVd trial to help mitigate ARs – 65% of patients in the XVd group had a dose reduction and the median dose of XPOVIO in that group was 80 mg once weekly.5,7 Patients in my clinical practice typically get reduced from 100 mg to 60 mg once weekly and experience minimal tolerability issues at 60 mg. In an exploratory post-hoc analysis of the XVd trial, efficacy was maintained with XPOVIO dose reductions (Figure 2).7
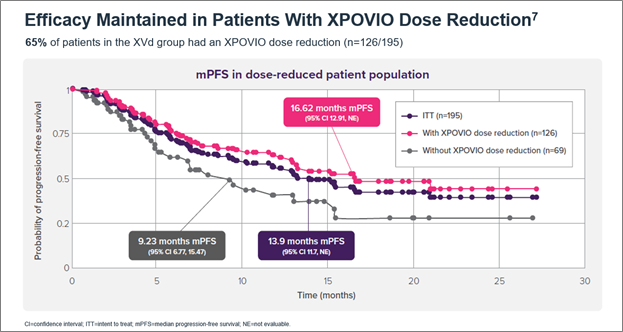 Figure 2. Median PFS in XPOVIO dose-reduced patients in the XVd trial.
Figure 2. Median PFS in XPOVIO dose-reduced patients in the XVd trial.
XVd was not associated with serious organ toxicities of the cardiac, pulmonary, renal, or hepatic systems.6,7 Warnings and precautions include life-threatening thrombocytopenia and neutropenia, gastrointestinal toxicities, severe life-threatening hyponatremia, serious infection, and life-threatening neurological toxicities.5 The most common adverse reactions (≥20% with a difference between arms of >5% compared to Vd) were fatigue, nausea, decreased appetite, diarrhea, peripheral neuropathy, upper respiratory tract infection, decreased weight, cataract, and vomiting (Figure 3).5 The XVd trial protocol required a prophylactic 5-HT3 antagonist to address nausea but allowed for other interventions as required.7 Nausea events were reported in 50% of patients, however, treatment-related nausea associated with XPOVIO diminished over time; 92% of nausea cases were resolved/resolving in the first month of treatment.7 Patients should be counseled on what to expect with XPOVIO therapy and monitored throughout treatment, with more frequent monitoring during the first three months of treatment.5
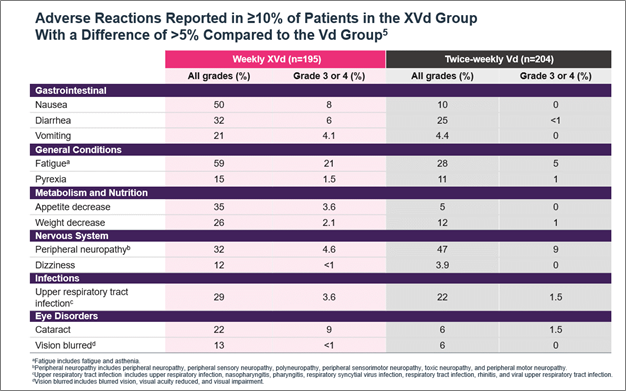 Figure 3. Adverse reactions reported in the XVd trial.
Figure 3. Adverse reactions reported in the XVd trial.
Below we consider 2 hypothetical patients where XPOVIO may be considered.
Patient A is a 66-year-old woman with relapsed/refractory MM. She was started on lenalidomide, bortezomib, and dexamethasone, and received autologous stem cell transplant (ASCT) followed by lenalidomide maintenance, which she did well on for 16 months. Upon relapsing, she was given daratumumab, pomalidomide with dexamethasone, and after 7 months, imaging confirmed that her MM progressed again. Given her DPd exposure, Patient A (RVd → ASCT → R → DPd) may be a candidate for a class switch to XVd.
Patient B is a 74-year-old man with a history of hypertension and was diagnosed with MM 2 years ago. Because of his hypertension, he was unable to start a PI due to risk of cardiotoxicity and he is ASCT ineligible. His healthcare provider started him on daratumumab, lenalidomide, and dexamethasone (DRd), but after 2 years, he has relapsed. A class switch to XPOVIO could be considered for Patient B as his second-line therapy.
Healthcare providers should consider patients’ individual clinical characteristics when making treatment decisions. Consider switching class with XPOVIO® (selinexor) for patients at relapse, including those who have been exposed to a CD38-mAb–based regimen.5 Based on the results of the XVd trial and considering the clonal nature of MM, switching patients to XPOVIO may be an option to consider.
INDICATIONS
XPOVIO® (selinexor) is a prescription medicine approved:
• in combination with bortezomib and dexamethasone to treat adult patients with multiple myeloma who have received at least one prior therapy.
• in combination with dexamethasone for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least four prior therapies and whose disease is refractory to at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti‐CD38 monoclonal antibody.
IMPORTANT SAFETY INFORMATION
Thrombocytopenia: XPOVIO can cause life-threatening thrombocytopenia, potentially leading to hemorrhage. Thrombocytopenia was reported in patients with multiple myeloma.
Thrombocytopenia is the leading cause of dosage modifications. Monitor platelet counts at baseline and throughout treatment. Monitor more frequently during the first 3 months of treatment. Monitor patients for signs and symptoms of bleeding. Interrupt, reduce dose, or permanently discontinue based on severity of adverse reaction.
Neutropenia: XPOVIO can cause life-threatening neutropenia, potentially increasing the risk of infection.
Monitor more frequently during the first 3 months of treatment. Consider supportive measures, including antimicrobials and growth factors (e.g., G-CSF). Interrupt, reduce dose, or permanently discontinue based on severity of adverse reaction.
Gastrointestinal Toxicity: XPOVIO can cause severe gastrointestinal toxicities in patients.
Nausea/Vomiting/Diarrhea: Provide prophylactic antiemetics or treatment as needed.
Anorexia/Weight Loss: Monitor weight, nutritional status, and volume status at baseline and throughout treatment and provide nutritional support, fluids, and electrolyte repletion as clinically indicated.
Hyponatremia: XPOVIO can cause severe or life-threatening hyponatremia.
Monitor sodium level at baseline and throughout treatment.
Serious Infection: XPOVIO can cause serious and fatal infections. Atypical infections reported after taking XPOVIO include, but are not limited to, fungal pneumonia and herpesvirus infection.
Neurological Toxicity: XPOVIO can cause life-threatening neurological toxicities.
Coadministration of XPOVIO with other products that cause dizziness or mental status changes may increase the risk of neurological toxicity.
Advise patients to refrain from driving and engaging in hazardous occupations or activities until the neurological toxicity fully resolves. Institute fall precautions as appropriate.
Embryo-Fetal Toxicity: XPOVIO can cause fetal harm when administered to a pregnant woman.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with a female partner of reproductive potential to use effective contraception during treatment with XPOVIO and for 1 week after the last dose.
Cataracts: New onset or exacerbation of cataract has occurred during treatment with XPOVIO. The incidence of new onset or worsening cataract requiring clinical intervention was reported.
ADVERSE REACTIONS
The most common adverse reactions (ARs) (≥20%) in patients with multiple myeloma who received XVd were fatigue, nausea, decreased appetite, diarrhea, peripheral neuropathy, upper respiratory tract infection, decreased weight, cataract, and vomiting.
Grade 3-4 laboratory abnormalities (≥10%) were thrombocytopenia, lymphopenia, hypophosphatemia, anemia, hyponatremia and neutropenia.
Fatal ARs occurred in 6% of patients within 30 days of last treatment. Serious ARs occurred in 52% of patients. Treatment discontinuation rate due to ARs was 19%. The most frequent ARs requiring permanent discontinuation in >2% of patients included fatigue, nausea, thrombocytopenia, decreased appetite, peripheral neuropathy and vomiting. Adverse reactions led to XPOVIO dose interruption in 83% of patients and dose reduction in 64% of patients.
USE IN SPECIFIC POPULATIONS
No overall difference in effectiveness of XPOVIO was observed in patients >65 years old when compared with younger patients. Patients ≥65 years old had a higher incidence of discontinuation due to an adverse reaction (AR) and a higher incidence of serious ARs than younger patients.
The effect of end-stage renal disease (CLCR <15 mL/min) or hemodialysis on XPOVIO pharmacokinetics is unknown.
Please see full Prescribing Information.
To report SUSPECTED ADVERSE REACTIONS, contact Karyopharm Therapeutics Inc. at 1-888-209-9326 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
© 2022 Karyopharm Therapeutics Inc. US-XPOV-10/22-00003
References
1. Mikkilineni L, Kochenderfer JN. CAR T cell therapies for patients with multiple myeloma. Nat Rev Clin Oncol. 2021;18(2):71-84. doi:10.1038/s41571-020-0427-6
2. Braunlin M, Belani R, Buchanan J, Wheeling T, Kim C. Trends in the multiple myeloma treatment landscape and survival: a U.S. analysis using 2011-2019 oncology clinic electronic health record data. Leuk Lymphoma. 2021;62(2):377-386. doi:10.1080/10428194.2020.1827253
3. Gandhi UH, Cornell RF, Lakshman A, et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33(9):2266-2275. doi:10.1038/s41375-019-0435-7
4. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Multiple Myeloma V.5.2022. © National Comprehensive Cancer Network, Inc. 2022. All rights reserved. Accessed October 18, 2022. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
5. XPOVIO (selinexor) [prescribing information]. Karyopharm Therapeutics Inc. https://www.karyopharm.com/wp-content/uploads/2019/07/NDA-212306-SN-0071-Prescribing-Information-01July2019.pdf
6. Grosicki S, Simonova M, Spicka I, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. The Lancet. 2020;396(10262):1563-1573. doi:10.1016/S0140-6736(20)32292-3
7. Data on File. Karyopharm Therapeutics Inc. 2021. Published online 2021.
